Définitions
L’AMP vigilance a pour objet la surveillance des incidents relatifs aux gamètes, aux tissus germinaux et aux embryons utilisés à des fins d’assistance médicale à la procréation, ainsi que des effets indésirables observés chez les donneurs de gamètes ou chez les personnes qui ont recours à l’assistance médicale à la procréation. Cette surveillance repose à ce jour sur un système de notifications spontanées.
On entend par incident tout accident ou erreur survenant au cours des différentes étapes du processus d’AMP, susceptible d’entraîner un effet indésirable sur la personne ou entrainant la perte de gamètes, de tissus germinaux ou d’embryons. À titre d’exemple, des incidents à type de perte totale d’embryons liée à un matériel défectueux (ex : panne de congélateur) ou de problèmes d’étiquetage des tubes de liquide folliculaire ont été rapportés.
On entend par effet indésirable toute réaction nocive survenant chez une personne, liée ou susceptible d’être liée aux activités d’AMP au cours de ses différentes étapes (stimulation, ponction folliculaire, insémination, transfert embryonnaire). À titre d’exemple, des effets indésirables à type d’hyperstimulations ovariennes sévères, d’hémopéritoine et d’accidents thromboemboliques ont été rapportés.
Dispositif d’AMP vigilance
L’AMP vigilance est une vigilance sanitaire réglementée confiée à l’Agence de la biomédecine par la loi n°2004-800 du 6 août 2004 relative à la bioéthique. Le décret de juin 20081 a complété l’organisation du dispositif et précisé les rôles des différents acteurs. La loi n°2011-814 du 7 juillet 2011 relative à la bioéthique n’a pas introduit de modifications dans ce champ.
Chaque centre autorisé pour les activités d’AMP doit désigner un correspondant local d’AMP vigilance (CLA). Ce dernier a pour missions de recueillir tous les incidents et les effets indésirables, de les déclarer sans délai à l’Agence de la biomédecine, d’informer les autres correspondants locaux d’AMP vigilance si nécessaire et les autres vigilances sanitaires concernées de son établissement, de participer aux investigations, d’aviser l’Agence de la biomédecine des résultats des investigations et en cas de difficultés de fonctionnement du dispositif. Tout autre professionnel qui ne dépend pas d’un centre d’AMP et qui a connaissance de la survenue d’un incident ou d’un effet indésirable doit en informer sans délai l’Agence de la biomédecine. En pratique, il est préférable que ce professionnel de santé en informe le centre d’AMP dans lequel le couple a été pris en charge initialement et que le CLA fasse la déclaration de l’événement indésirable.
Fin 2015, le réseau d’AMP vigilance comprenait 186 correspondants locaux désignés dans 100 % des centres d’AMP autorisés.
Les déclarations d’AMP vigilance sont évaluées et expertisées par l’Agence de la biomédecine en lien avec un groupe de travail externe “AMP vigilance” composé d’experts biologistes et gynécologues-obstétriciens spécialisés dans le domaine de l’AMP. L’Agence de la biomédecine a la possibilité de proposer ou de mettre en œuvre des mesures préventives ou correctives dans un but de réduction des risques et d’amélioration des pratiques. L’Agence de la biomédecine assure le secrétariat de la commission nationale d’AMP vigilance.
L’AMP vigilance comporte une dimension transversale forte, impliquant souvent d’autres systèmes de vigilance sanitaire, notamment la matériovigilance et la pharmacovigilance. Ceci a justifié le développement de procédures d’échanges des données avec ces systèmes de vigilance sanitaire pilotés par l’ANSM.
Matériel et méthodes
Les incidents et les effets indésirables doivent faire l’objet d’une déclaration par les CLA à l’Agence de la biomédecine au moyen d’une fiche, de préférence par l’application informatique AMP Vigie.
En 2015, 97% des déclarations ont été saisies directement dans AMP Vigie par le CLA du centre d’AMP
Les incidents et effets indésirables recueillis sont analysés et évalués en termes de gravité et d’imputabilité.
On distingue ainsi 5 niveaux de gravité, de G1 à G5, présentés ci-dessous :
| G1 | Diminution de la performance du processus, sans conséquence sur son résultat, et/ou source de contrainte opérationnelle acceptable |
| G2 | Dégradation de la performance du processus susceptible ou ayant altéré de façon modérée son résultat et/ou source de contrainte opérationnelle non acceptable Perte d’embryons et/ou de gamètes sans disparition des chances de procréation sur la tentative |
| G3 | Dégradation de la performance du processus ayant altéré de façon importante son résultat. Complications liées au processus d’AMP avec hospitalisation et/ou incapacité fonctionnelle mineure Intervention médicale ou chirurgicale afin d’exclure tout dommage permanent ou infirmité corporelle Risque de transmission d’affection(s) à morbidité modérée accessible(s) à un traitement Perte d’embryons et/ou des gamètes avec disparition des chances de procréation sur la tentative |
| G4 | Acte ou procédure sur un patient autre (erreur d’attribution) Perte d’embryons et/ou des gamètes avec disparition définitive des chances de procréation pour le couple Complications sévères liées au processus d’AMP avec hospitalisation supérieure à 7 jours et/ou incapacité fonctionnelle majeure Risque de transmission par les gamètes d’affection(s) à morbidité sévère : affections transmissibles avec mise en jeu du pronostic vital |
| G5 | Décès au cours du processus d’AMP Incapacité fonctionnelle majeure et permanente |
L’imputabilité est une estimation individuelle, pour une déclaration donnée, de la probabilité de la relation existante entre le processus d’AMP et la survenue d’un effet indésirable. L’ensemble des étapes du processus d’AMP et leur environnement doivent être pris en compte. Il peut y avoir une différence entre le niveau d’imputabilité établi lors de la survenue de l’événement indésirable et celui retenu après investigation du cas. Il s’agit donc d’une estimation initiale qui est réévaluée et modifiée si besoin par le CLA dans la partie B de la fiche de déclaration. Après réception de chaque déclaration, l’Agence de la biomédecine réévalue son niveau d’imputabilité pour s’assurer d’une utilisation cohérente de l’échelle ou attribue une imputabilité à cet événement si cela n’a pas déjà été fait par le déclarant.
Résultats
I. Données générales
En 2015, l’Agence de la biomédecine a reçu un total de 489 déclarations d’AMP vigilance2. Celles-ci provenaient de 83 centres d’AMP (80 centres clinico-biologiques et 3 laboratoires d’insémination artificielle), ce qui représente en moyenne 6 déclarations ± 5 par centre [1-29].
De plus, onze notifications provenant du système d’alerte européen « RATC » et concernant essentiellement la découverte de maladies génétiques chez des enfants nés à l’étranger de femme ayant eu recours à un don de sperme provenant d’une banque étrangère ont également été réceptionnées en AMP-vigilance. Ces notifications, bien que ne faisant pas partie stricto sensu du champ de la vigilance, sont transmises à l’ABM dès lors qu’une ou plusieurs femmes françaises sont a priori concernées par l’utilisation des paillettes de sperme du donneur incriminé.
Ces 489 déclarations sont réparties en 366 effets indésirables et en 125 incidents, ce qui correspond à 491 événements indésirables car deux déclarations concernaient à la fois un incident et un effet indésirable liés.
Parmi ces déclarations, 24% ont fait l’objet d’une déclaration à au moins une autre vigilance. Ce qui reflète bien la dimension transversale importante de l’AMP vigilance et la nécessité d’une coordination avec les autres systèmes de vigilance que ce soit au niveau de l’établissement ou au niveau national.
Nombre de déclaration
L’évolution du nombre de déclarations d'AMP vigilance et du nombre de centres déclarants est présentée à la figure FAMPV1.
Figure FAMPV1. Evolution du nombre de déclarations d’AMP vigilance et du nombre de centres déclarants de 2011 à 2015
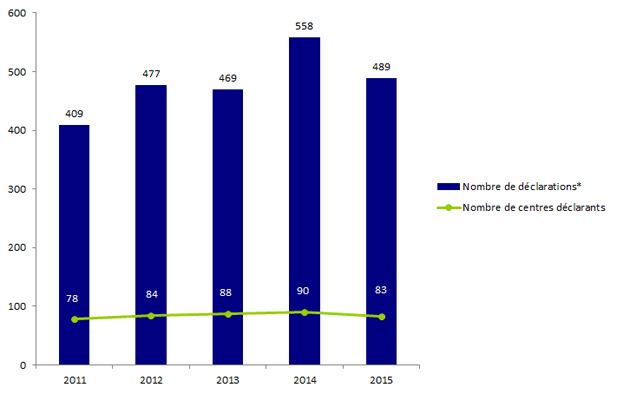
* Déclarations d'événements indésirables adressées à l'Agence de la biomédecine (quelle que soit l'année de constatation de l'événement)
L’analyse du nombre de déclarations par année est un indicateur qui permet d’apprécier en partie l’efficacité du dispositif mis en place. Le nombre de déclarations a pratiquement triplé depuis la mise en place du dispositif en 2008, néanmoins on observe un tassement entre 2014 et 2015, avec une baisse des déclarations de l’ordre de 12%.
Depuis 2009, tous les centres clinico-biologiques d’AMP et près de vingt centres d’insémination ont fait au moins une déclaration en AMP vigilance.
En moyenne, l’Agence de la biomédecine a reçu 41 déclarations par mois, avec un pic de 58 déclarations en juillet et de 87 déclarations en décembre.
Délai de déclaration
Le décret de juin 2008 prévoit que les CLA déclarent les incidents et les effets indésirables sans délai à l’Agence de la biomédecine ce qui, en pratique, se traduit par un délai de déclaration ne devant pas excéder 48 heures pour les événements graves.
Le délai moyen entre la déclaration faite à l’Agence de la biomédecine en 2015 et la date de constatation de l’événement indésirable est bien supérieur avec une moyenne de 53 jours et une médiane à 27 jours. Il existe une grande variabilité de ce délai attestée par la valeur de l’écart-type à 78 jours et des valeurs extrêmes comprises entre 0 et 524 jours.
La répartition et l’évolution du délai de déclaration depuis 2011 est présentée dans le tableau TAMPV1.
Tableau TAMPV1. Répartition des événements indésirables selon le délai de déclaration
Année de la déclaration |
Délai* |
Total |
|||
≤ 1 mois** |
] 1 mois - 6 mois] |
] 6 mois - 12 mois] |
≥ 12 mois |
||
2011 |
36,4% |
49,1% |
13,4% |
1,0% |
100% (409) |
2012 |
36,5% |
49,9% |
10,7% |
2,9% |
100% (477) |
2013 |
45,4% |
46,9% |
6,8% |
0,9% |
100% (469) |
2014 |
44,4% |
46,9% |
7,7% |
1,1% |
100% (559) |
2015 |
54,8% |
39,1% |
4,5% |
1,6% |
100% (491) |
* Délai en jours mesuré entre la date à laquelle l'événement indésirable est constaté et la date de la déclaration de cet événement à l'Agence de la biomédecine
** 31 jours
En 2015, plus de la moitié des déclarations ont été envoyées dans le mois qui suit la constatation de l’événement indésirable. Néanmoins, près de 6% des déclarations ont été effectuées plus de 6 mois après la constatation de l’événement.
Afin de favoriser une meilleure réactivité pour l’envoi de la fiche initiale de déclaration, une réflexion est en cours afin de simplifier la partie A de la fiche qui contiendra essentiellement des données factuelles et descriptives de l’événement constaté. Le résultat des investigations et le descriptif des mesures correctives ou préventives éventuelles seront renseignés secondairement dans la partie B.
II. Effets indésirables
Parmi les 489 déclarations d’événements indésirables, l’Agence de la biomédecine a reçu 366 effets indésirables dont 93,2% étaient des effets graves (n=341), c’est-à-dire susceptibles d’entraîner la mort ou de mettre la vie en danger, d’entraîner une invalidité ou une incapacité, de provoquer ou de prolonger une hospitalisation ou tout autre état morbide ou susceptibles de se reproduire chez un ou plusieurs donneurs ou personnes qui ont recours à l’AMP.
Taux de notification des effets indésirables par classe d’activité
L’évolution du nombre de déclarations d’effets indésirables selon les classes d’activité (AMP, Autoconservation, Dons) est présentée dans le tableau TAMPV2.
Tableau TAMPV2. Nombre de déclarations d’effets indésirables selon les classes d’activité (AMP, Autoconservation, Dons)
Activité |
2013 |
2014 |
2015 |
||
Nombre d'actes d'AMP(a) |
Nombre d'effets indésirables |
Nombre d'actes d'AMP(a) |
Nombre d'effets indésirables |
Nombre d'effets indésirables |
|
AMP |
142771 |
361 |
143778 |
420 |
357 |
Inséminations |
57352 |
4 |
56468 |
6 |
6 |
Fécondations (FIV, ICSI) |
62364 |
337 |
61894 |
386 |
326 |
TEC |
23055 |
3 |
25416 |
5 |
7 |
DPI |
628 |
8 |
905 |
6 |
6 |
Type d'AMP non renseigné |
0 |
12 |
0 |
19 |
12 |
Autoconservations |
11131 |
0 |
11709 |
1 |
3 |
Préservation de la fertilité(b) |
4882 |
. |
5313 |
. |
. |
En cours d'AMP(c) |
6249 |
. |
6396 |
. |
. |
Dons (gestion des donneurs) |
757 |
1 |
739 |
2 |
6 |
Donneurs de spermes |
303 |
1 |
238 |
0 |
0 |
Donneurs d'ovocytes |
454 |
0 |
501 |
2 |
6 |
- (a) Tentatives : cycles d'insémination artificielle (IIU, IIC) ; ponctions d'ovocytes dans le cadre des fécondations in vitro (FIV, ICSI) ; transferts d'embryons congelés (TEC)
- (b) Il s’agit des nouvelles autoconservations de spermatozoïdes réalisées dans l’année (en nombre de patients)
- (c) Il s’agit des nouvelles autoconservations de l’année (en nombre de patients). Les autoconservations concernent les tissus germinaux quel qu’il soit (spermatozoïdes, ovocytes, tissus testiculaires et ovariens
En AMP, en moyenne, au niveau national, si on rapporte le nombre d’effets indésirables déclarés à l’activité, on obtient un indicateur de 2,53 effets indésirables déclarés pour 1000 actes en 2013 (rapporté à l’activité 2013), de 2,92‰ en 2014 (rapporté à l’activité de 2014) et de 2,48‰ en 2015 (rapporté à l’activité de 2014). La légère diminution de cet indicateur en 2015 est retrouvée pour toutes les activités, soit en raison d’un moindre nombre d’événements déclarés en 2015 (cas des activités de fécondations), soit en lien avec une augmentation du nombre d’actes (cas des activités de TEC et de DPI).
Répartition des effets indésirables selon la typologie
La distribution des effets indésirables selon la typologie en 2015 est présentée à la figure FAMPV2.
Figure FAMPV2. Répartition des types d’effets indésirables (n=366)
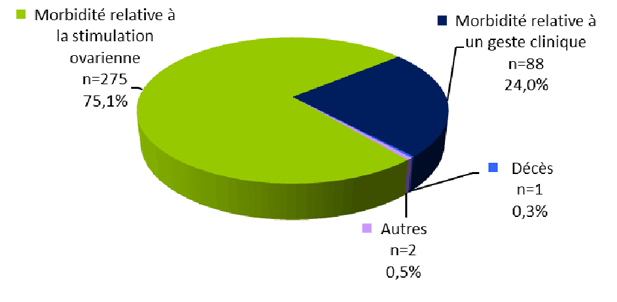
Pour l’année 2015, parmi les 366 effets indésirables déclarés : 275 (75%) concernent la stimulation ovarienne, 88 (24%) concernent un geste clinique lors de l’AMP (inséminations, ponctions, transferts), et 2 (0,5%) sont classés comme « autres ».
Il est à souligner la déclaration du décès d’une patiente d'origine mauritanienne après son retour dans son pays et 40 jours après une tentative de FIV-ICSI avec transfert d'embryons. Aucune information clinique sur les circonstances du décès n’a pu être obtenue malgré une demande écrite auprès du médecin concerné ainsi qu'auprès du directeur de l'établissement où a eu lieu le décès.
- Effets indésirables relatifs à la stimulation ovarienne
La distribution des effets indésirables relatifs à la stimulation ovarienne selon leur gravité est présentée à la figure FAMPV3
Figure FAMPV3. Répartition des effets indésirables relatifs à la stimulation ovarienne selon leur gravité (n=275)
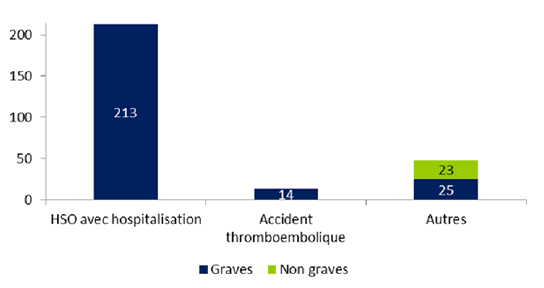
* HSO avec hospitalisation hors accidents thromboemboliques et torsions d’annexes
Soixante-quinze pour cent des effets indésirables rapportés concernent des événements relatifs à la stimulation ovarienne (n = 275). Ces 275 effets indésirables comprennent essentiellement des effets indésirables graves (92%). Il s’agit le plus souvent de syndromes d’hyperstimulation ovarienne sévères avec hospitalisation d’au moins 24 heures (213/275 soit 77%). Quatorze cas d’accidents thromboemboliques ont été rapportés par 11 centres d’AMP chez des femmes âgées en moyenne de 33 ± 3 ans [29-40]. Ces 14 cas sont répartis en :
- 5 embolies pulmonaires ;
- 5 phlébites des membres inférieurs ;
- 1 thrombose de la veine porte ;
- 2 accidents vasculaires cérébraux ;
- 1 phlébite jugulaire et du tronc brachiocéphalique.
Dans 3 observations, il est mentionné un syndrome d’hyperstimulation ovarienne sévère associé.
Parmi les 48 événements renseignés comme « Autres », on retrouve 29% de torsions d’annexes (14/48) et 46% d’HSO sans hospitalisation (22/48).
La part importante des déclarations relatives aux syndromes d’hyperstimulation ovarienne, qui atteint cette année 65% des déclarations mais dont le nombre a baissé en valeur absolue, a motivé la mise en ligne d’un outil évaluation des pratiques professionnelles par l’Agence de la biomédecine (https://www.eppshos.org/). Cet outil est accessible aux professionnels et un rappel en ce sens leur a été adressé par courrier électronique en mars 2015.
- Effets indésirables relatifs à un geste clinique
La distribution des effets indésirables relatifs à un geste clinique lors de l’AMP selon leur gravité est présentée à la figure FAMPV4.
Figure FAMPV4. Répartition des effets indésirables relatifs à un geste clinique selon leur gravité (n=88)
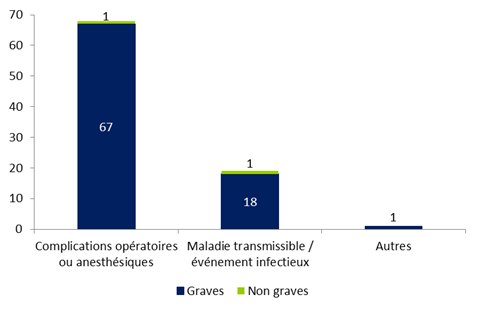
En 2015, 88 effets indésirables (24%) sont en lien avec un geste clinique lors de l’AMP (insémination, ponction folliculaire, transfert embryonnaire, ...). Ces effets indésirables sont principalement des effets graves (86/88). Parmi ces événements, 77% (68/88) concernent des complications chirurgicales telles que des hémopéritoines (n=50), des hématomes ovariens, des douleurs pelviennes ou des complications anesthésiques dont 1 cas de probable passage intravasculaire de ropivacaïne ayant entraîné une crise convulsive suivie d'un arrêt cardiaque (avec récupération sans séquelle). Il est à noter que le retour d’expérience de l’Agence de la biomédecine du 1er février 2013 concernant les accidents anesthésiques lors des ponctions ovariennes a été ré-adressé au centre pour modification de leur procédure.
Gravité / Conséquences
La gestion des déclarations par l’Agence de la biomédecine est notamment basée sur le niveau de gravité des effets indésirables rapportés.
Il existe 5 niveaux de gravité allant de G1 à G5, les niveaux G3 à G5 correspondant à des événements indésirables avec des conséquences graves.
La distribution des effets indésirables en 2015 en fonction de la gravité est présentée dans la figure FAMPV5.
Figure FAMPV5. Gravité des effets indésirables en 2015
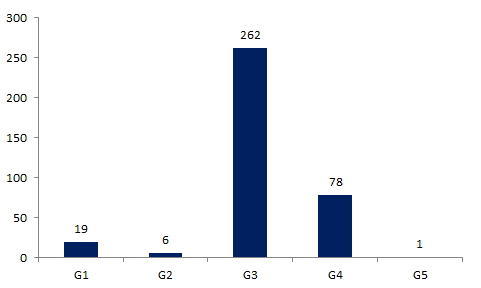
Les effets indésirables graves (gravité G3 et au-delà) représentent 93,2% (341) des événements indésirables rapportés. Un effet indésirable est considéré comme grave s’il est susceptible d’entraîner la mort ou de mettre la vie en danger, d’entraîner une invalidité ou une incapacité, de provoquer ou de prolonger une hospitalisation ou tout autre état morbide ou susceptible de se reproduire chez un ou plusieurs donneurs ou personnes qui ont recours à l’AMP. Cette proportion importante d’effets indésirables graves est directement liée au nombre important d’HSO avec hospitalisation déclarées.
Le type de conséquences pour les 366 effets indésirables observés chez le patient est présenté au tableau TAMPV3.
Tableau TAMPV3. Conséquences des effets indésirables chez les couples ou patients concernés
|
Conséquences |
||
|
N |
Incapacité |
Décès |
Hospitalisation et mise en jeu du pronostic vital |
22 |
1 |
0 |
Hospitalisation sans mise en jeu du pronostic vital |
303 |
22 |
0 |
Aucune hospitalisation |
40 |
0 |
0 |
Inconnue* |
1 |
0 |
1 |
Total (événements chez le patient) |
366 |
23 |
1 |
Parmi les 366 déclarations d’effets indésirables, la plupart (325/366 soit 89%) ont entraîné une hospitalisation. Pour 1 déclaration, cette information n’a pas été renseignée. Parmi ces 325 cas d’hospitalisation, 7% (22/325) ont été déclarés comme pouvant mettre en jeu le pronostic vital avec pour conséquences dans 1 cas une incapacité/invalidité.
Les durées d’hospitalisation en fonction du type d’effet indésirable sont présentées dans le tableau TAMPV4.
Tableau TAMPV4. Durée d'hospitalisation (jours) selon le type d'effet indésirable
Typologie |
Sous-typologie |
N* |
Moyenne |
Ecart-type |
Médiane |
Evénements relatifs à une stimulation ovarienne |
HSO avec hospitalisation |
203 |
6,1 |
4,2 |
5 |
Accident thromboembolique |
9 |
8,2 |
6,1 |
8 |
|
Autres |
24 |
2,3 |
1,0 |
2 |
|
Evénements relatifs à un geste clinique |
Complications opératoires ou anesthésiques |
53 |
3,2 |
2,2 |
3 |
Maladie transmissible / événement infectieux |
18 |
6,8 |
5,7 |
6 |
|
Autres |
2 |
2,5 |
2,1 |
2.5 |
|
Autres |
1 |
5,0 |
NC |
5 |
|
Total |
310 |
5,4 |
4,2 |
4 |
|
*Nombre d’effets indésirables pour lesquels la durée d’hospitalisation a été renseignée (non renseignée : n=15)
NC : non calculable
Parmi les 325 déclarations d’effets indésirables ayant entraîné une hospitalisation, la durée d’hospitalisation a été renseignée pour 310 effets. Pour ces effets, la durée moyenne d’hospitalisation a été d’environ 5,4 jours avec une médiane à 4 jours. Les durées moyennes d’hospitalisation les plus longues s’observent dans les cas d’accidents thromboemboliques et les complications infectieuses.
Parmi les 203 HSO avec durée d’hospitalisation mentionnée, 5 ont entrainé une hospitalisation d'une journée et 198 une hospitalisation de plus d'une journée. La durée d’hospitalisation la plus longue a été de 29 jours chez une patiente de 35 ans pour une HSO après transfert embryonnaire avec 5 jours de réanimation puis séjour dans le service de gynécologie jusqu’à la normalisation biologique (cytolyse) et clinique (ascite).
D’après les données enregistrées dans l’outil AMP-vigie, ces effets indésirables ont généré de façon cumulée un nombre total de 1 675 journées d’hospitalisation.
Actions entreprises par le centre
L’analyse des données concernant les déclarations d’effets indésirables met en évidence une relative confusion par les CLA entre les informations relatives à la prise en charge des patients concernés par l’événement (ex : hospitalisation de la patiente, administration d’un antibiotique ou d’un antalgique, etc.) et la mise en œuvre de mesures correctives destinées à limiter le risque de récurrence ou les conséquences potentielles des effets indésirables (modification de procédures, formation du personnel, acquisition de matériel, etc.). Une action pédagogique devra être initiée par l’Agence de la biomédecine afin de mieux distinguer ces éléments dans l’objectif de souligner, en ce qui concerne la vigilance, l’importance des mesures correctives ou préventives.
En 2015, hors prise en charge thérapeutique, le type d’actions mises en œuvre par le centre suite à la survenue d’un effet indésirable est présenté dans le tableau AMPV5.
Tableau TAMPV5. Actions entreprises par les centres
Type d'action |
N |
|
Signalement à une autre vigilance par le centre* |
Pharmacovigilance |
68 |
Hémovigilance |
1 |
|
Biovigilance |
0 |
|
Matériovigilance |
0 |
|
Autres |
7 |
|
Total |
76 |
|
Mesures préventives concernant l'organisation** |
Réparation ou remplacement du matériel défectueux |
1 |
Mise en place ou modification de procédure |
15 |
|
Autres |
2 |
|
Total |
18 |
|
* 73 effets indésirables ont fait l’objet d’un signalement à au moins une autre vigilance (2 des effets indésirables ont été signalés à la fois à la pharmacovigilance et à un autre système de vigilance, et 1 effet a indésirable a été signalé à la fois à la pharmacovigilance et l’hémovigilance) (13 centres)
** 15 centres ont mis en place des mesures préventives concernant l’organisation à la suite d’un effet indésirable (18 déclarations), 6 déclarations (5 centres) ont été signalées à une au moins une autre vigilance et ont été à l’origine de mesures préventives concernant l’organisation.
Activité de don ou événement donneur
Parmi les 366 effets indésirables, 6 cas concernaient des donneuses d’ovocytes. Ces événements sont peu fréquents (inférieur à 1%) et leur répartition depuis 2011 est présentée dans le tableau AMPV6.
Tableau TAMPV6. Evolution depuis 2011 des effets indésirables « donneurs »
|
2011 |
2012 |
2013 |
2014 |
2015 |
|
Stimulation ovarienne |
HSO avec hospitalisation |
0 |
2 |
1 |
1 |
2 |
Autres |
0 |
0 |
0 |
0 |
1 |
|
Geste clinique |
Complications opératoires ou anesthésiques |
2 |
1 |
0 |
1 |
3 |
Maladie transmissible / événement infectieux |
0 |
1 |
0 |
0 |
0 |
|
Total |
2 |
4 |
1 |
2 |
6 |
|
Le détail des 6 déclarations réceptionnées en 2015 est résumé ci-après :
- un choc hémorragique sur hémopéritoine post-ponction ovocytaire ayant nécessité une reprise chirurgicale per-coelioscopique pour électro-coagulation et 10 jours d’hospitalisation (à noter, la mise en œuvre d’une mesure correctrice préventive consistant en la réalisation systématique d’un contrôle échographique lors de la survenue d’une évolution clinique inhabituelle pendant la phase de surveillance post-ponction) ;
- un hémopéritoine post-ponction ovocytaire ayant nécessité une reprise chirurgicale per-coelioscopique et 3 jours d’hospitalisation ;
- un hémopéritoine post-ponction ovocytaire ayant nécessité une reprise chirurgicale per-coelioscopique et 2 jours d’hospitalisation ;
- une torsion d'annexe 10 jours après la ponction ovarienne ayant nécessité une coelioscopie en urgence pour détorsion avec suite simple ;
- une hyperstimulation ovarienne modérée sans complication associée chez une donneuse de 28 ans ayant entrainée une hospitalisation de 4 jours ;
- une hyperstimulation ovarienne modérée sans complication associée chez une donneuse de 33 ans ayant entrainée une hospitalisation de 3 jours.
Parmi ces 6 déclarations, toutes ont été considérées comme sévères soit en raison de la typologie de l’événement (choc, hémopéritoine, torsion d’annexe), soit en raison de la durée d’hospitalisation des patientes (cas notamment des 2 déclarations d’hypertimulation ovarienne). Concernant les hyperstimulations ovariennes, l’hospitalisation des patientes a essentiellement été justifiée par la mise en œuvre d’une surveillance clinico-biologique. Il ne s’agit donc pas stricto sensu d’effets indésirables sévères. En excluant ces 2 cas ou en les prenant en considération, les taux de complications chez les donneuses d’ovocytes (taux de 0,54% et taux de 0,81% respectivement) restent comparables aux données de la littérature scientifique3 qui font mention de la survenue de complications sévères allant de 0,11% à 1,03% avec une moyenne à 0,7% et de complications mineures (ayant justifiées une consultation médicale) pouvant aller jusqu’à 8,5%.
Activité d’autoconservation
Parmi les 366 effets indésirables, 3 cas concernaient les activités d’autoconservation. Ces 3 effets indésirables concernaient :
- un hémopéritoine massif post-ponction ovocytaire ayant nécessité une reprise chirurgicale per-coelioscopique avec ovariectomie unilatérale et 3 jours d’hospitalisation ;
- un hémopéritoine post-ponction chez une patiente sous anticoagulant à dose curative ayant nécessité 4 jours d’hospitalisation ;
- une hyperstimulation ovarienne sur un syndrome des ovaires polykystiques ayant nécessité 5 jours d’hospitalisation.
II. Incidents
Parmi les 489 déclarations d’événements indésirables, l’Agence a reçu 125 incidents dont 28,8% étaient des incidents graves (n=36), c’est-à-dire susceptible d’entraîner des effets indésirables graves ou occasionnant une erreur d’attribution ou une perte de gamètes, tissus germinaux ou embryons avec perte de chance totale de procréation sur la tentative ou pour le couple.
Répartition des incidents par étape du processus
Afin d’identifier les étapes du processus les plus à risque d’incidents, la répartition des incidents par étape du processus et par gravité est représentée à la figure FAMPV6.
Figure FAMPV6. Répartition du nombre d’incidents par étape et par gravité
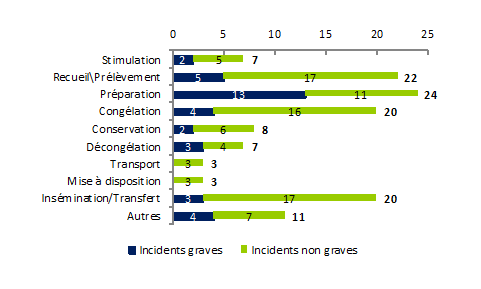
Bien que le nombre de déclarations ne soit pas exhaustif, ce graphe montre que les 4 étapes du processus qui semblent le plus à risque d’incidents sont :
- Le recueil/prélèvement des gamètes (18%)
- La préparation des gamètes/embryons (19%)
- La congélation des gamètes/embryons (16%)
- L’insémination/transfert (16%)
Parmi les incidents survenus lors de la préparation, environ 60% concernaient des incidents graves, c’est-à-dire pour lesquels l’incident a eu pour conséquence une perte totale des gamètes ou embryons sur la tentative. Inversement, lors de l’étape de stimulation ovarienne et de conservation, il s’agit plus particulièrement d’incidents non graves (respectivement 88% et 59%). Les étapes qui ont été renseignées comme « Autres » correspondaient notamment à la réception des gamètes, aux accords administratifs (décongélation, transfert d’embryon), à la conservation informatique des données et au bilan biologique pré - AMP.
Répartition des incidents selon la typologie
La répartition des incidents selon leur typologie est représentée dans la figure FAMPV7.
Figure FAMPV7. Répartition des incidents selon leur typologie (n=125)
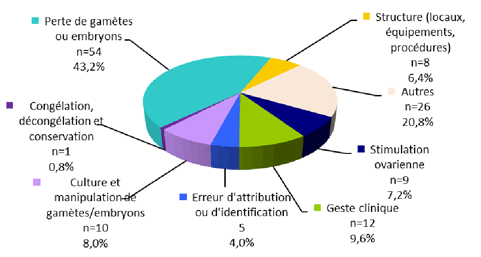
Les incidents classés comme « Autres » (26 soit 20,8%) concernaient :
- Le non-respect des obligations réglementaires inhérent aux couples (problème d’identification, fraude, etc.) ;
- Les autres typologies en lien avec les gamètes, embryons ou tissus germinaux telles la destruction accidentelle d’embryons en cours d’incubation, une erreur sur le choix de l’embryon à transférer parmi l’ensemble des embryons du couple ou bien le non-respect du circuit risque viral ;
- Les typologies « autres » proprement dites telles des fusions de dossiers dans un logiciel de traitement informatisé des données, une incompatibilité des versions du logiciel de traitement informatisé des données entre différents centres empêchant le transfert des dossiers ou une panne de la centrale de traitement d’air de la salle de préparation
- Incidents relatifs à la stimulation ovarienne
La distribution des incidents relatifs à la stimulation ovarienne selon leur gravité en 2015 est présentée à la figure FAMPV8.
Figure FAMPV8. Répartition des incidents relatifs à une stimulation ovarienne selon leur gravité (n=9)
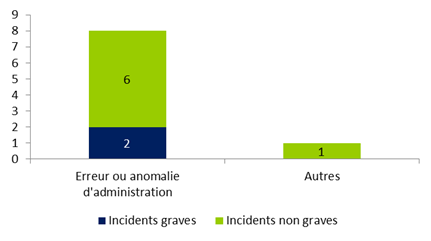
Parmi les 9 incidents déclarés concernant la stimulation ovarienne, plus de 88% (8/9) concernent des erreurs ou anomalies d’administration de traitement (erreur de dose, erreur de jour, erreur de médicament) qui sont pour la plupart des incidents non graves. L’autre incident concernait une erreur de transmission des résultats du dosage de la β-HCG.
- Incidents relatifs à la culture et préparation des embryons/gamètes
La distribution des incidents relatifs à la culture et préparation des embryons/gamètes selon leur gravité est présentée à la figure FAMPV9.
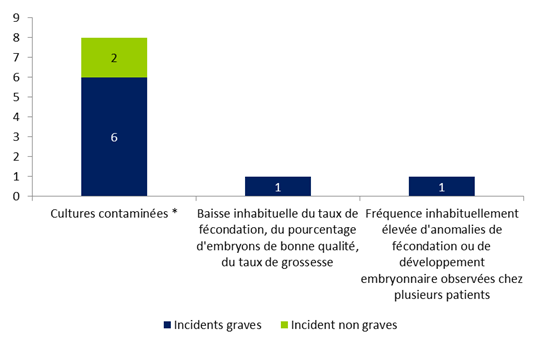
Les incidents lors de la culture et la préparation des gamètes ou des embryons représentent 8% des déclarations d’incidents et sont majoritairement des incidents graves (80%) car ils ont entrainé une perte totale des gamètes ou des embryons sur la tentative.
Parmi ces incidents, 80% (8/10) concernent des cultures contaminées qui pour la plupart sont des incidents graves. Plus de 60% de ces contaminations étaient d’origine spermatique (5/8) et les autres d’origine environnementale (1/8), manuportée (1/8) ou liée à la patiente (1/8).
Les deux autres incidents relatifs à la culture concernent, pour l’un, une fréquence inhabituellement élevée d’altération des embryons (taux de triploïdie anormal) possiblement en lien avec une modification du transfert thermique entre la platine chauffante et un nouveau lot de boîtes de culture et, pour l’autre, une baisse inhabituelle du taux de grossesses lors des inséminations possiblement en lien avec l’utilisation d’un nouveau lot de pipettes dont le procédé de stérilisation avait été modifié par le fabricant sans information des utilisateurs (passage à l’oxyde d’éthylène au lieu d’une irradiation par faisceau ionisant). Ces deux déclarations ont également fait l’objet d’une transmission en matériovigilance afin de disposer des résultats des investigations des fabricants. Concernant l’incident avec les pipettes compte-gouttes, une enquête a été réalisée par l’Agence de la biomédecine auprès des centres. Dix-huit centres ont répondu et parmi eux, 15 n’utilisaient pas ce type de matériel ou n’utilisaient pas cette référence de pipette. Deux centres utilisaient cette référence de pipettes mais avec d’autres lots stérilisés par irradiation. Enfin, un centre a signalé utiliser ces pipettes pour le traitement du sperme en insémination mais aussi pour les tests de migration survie des spermatozoïdes. Le lot concerné, stérilisé par l’oxyde d’éthylène n'avait pas encore été utilisé par ce centre qui disposait d’un autre lot, également stérilisé à l’oxyde d’éthylène, et dont l’usage avait été validé par la réalisation de tests de survie à 24 heures en laissant la préparation de sperme dans le compte-goutte sans observation de différence avec les préparations laissées classiquement en tubes. Enfin, le fabricant a adressé un courrier à l’ensemble de ces clients utilisateurs pour les informer du changement de procédé de stérilisation.
Figure FAMPV9. Répartition des incidents relatifs à la culture et préparation des embryons / gamètes selon leur gravité (n=10)
- Incidents relatifs à la perte ou à la destruction de gamètes/embryons
La distribution des incidents relatifs à une perte ou à une destruction de gamètes/embryons est présentée dans le tableau TAMPV7.
Tableau TAMPV7. Répartition des incidents relatifs à la perte ou la destruction des embryons / gamètes selon leur gravité (n=54)
|
Incidents non graves |
Incidents graves |
Total |
Maladresse ou incident lié à un environnement ou à un matériel particulier |
27 |
5 |
32 |
Conservation ou décongélation inappropriée |
4 |
3 |
7 |
Transport inapproprié |
1 |
0 |
1 |
Equipement défectueux |
5 |
3 |
8 |
Perte de paillettes ou rupture de paillettes |
3 |
0 |
3 |
Perte de traçabilité |
1 |
0 |
1 |
Acte de malveillance |
1 |
0 |
1 |
Autres |
1 |
0 |
1 |
Total |
43 |
11 |
54 |
Cinquante-quatre incidents (43%) rapportés en 2015 concernent des pertes ou des destructions accidentelles, partielles ou totales, de gamètes ou d’embryons. Il s’agit le plus souvent d’incidents non graves (79%). Dans trois-quarts des cas, ces incidents sont en rapport soit avec une difficulté ou une erreur liée à la gestuelle technique, soit avec un équipement ou un matériel défectueux. Pour rappel, une perte totale des gamètes ou embryons sur la tentative est considéré comme un incident grave.
Pour illustrer cette problématique, l’Agence de la biomédecine a réceptionné une déclaration faisant état de la remontée en température d’un récipient cryogénique à remplissage manuel contenant des ovocytes vitrifiés provenant de patientes prises en charge pour préservation de la fertilité. Cette déclaration, parvenue également en matériovigilance, a motivé l’envoi d’un courrier par l’Agence nationale de sécurité du médicament et des produits de santé à l’ensemble des utilisateurs. Par ailleurs, une sensibilisation des acteurs en AMP a été faite par l’Agence de la biomédecine via la diffusion dans le bulletin d’AMP-vigilance d’un focus sur les mesures à mettre en place pour réduire les risques de défaillance matérielle lors de l’utilisation de récipients à remplissage manuel.
Les préconisations émises afin de mieux sécuriser les échantillons biologiques conservés dans de tels équipements sont résumées ci-après :
1- Avant toute opération de remplissage, il est recommandé d’évaluer la quantité d’azote liquide s’étant évaporée depuis le précédent remplissage. Ce contrôle doit faire l’objet d’une traçabilité et toute évaporation excessive doit être signalée à la personne responsable du centre ;
2- Les opérations de remplissage doivent éviter tout choc sur l’enveloppe interne et notamment au niveau du col du récipient ;
3- Le niveau d’azote liquide ne doit pas dépasser le seuil maximal établi par le fabricant sous peine d’endommager le col du récipient. En conséquence, il ne faut pas remplir les récipients à « bouchon flottant ».
4- Pour éviter le dépassement du seuil haut, il est important de contrôler le niveau d’azote lors des opérations de remplissage du récipient et, à cette fin, il est recommandé de diminuer le débit pour éviter tout bouillonnement et formation de vapeur pouvant gêner le contrôle visuel ;
5- Toute observation d’anomalie, telles des altérations de la peinture du revêtement extérieur ou des zones de condensation, peut être le témoin d’un mésusage ou d’une perte des caractéristiques d’isolation du récipient et devra être signalée à la personne responsable ;
6- En cas d’anomalies constatées ou d’évaporation excessive, il peut être nécessaire de transférer le contenu du récipient vers un récipient de secours afin de pouvoir procéder à sa vérification par le fournisseur ;
7- Enfin, il peut être également utile d’équiper les récipients cryogéniques d’une alimentation automatisée et de contrôles de niveau et de température avec report des alarmes.
Un groupe de travail sur ce sujet sera mis en place en 2016 et les recommandations qui en seront issues pourront être intégrées, si nécessaire, dans les règles de bonnes pratiques en AMP.
- Incidents relatifs à la structure (locaux, équipements, procédures)
La répartition des incidents relatifs à la structure selon leur gravité est présentée à la figure FAMPV10.
Figure FAMPV10. Répartition des incidents relatifs à la structure selon leur gravité (n=8)
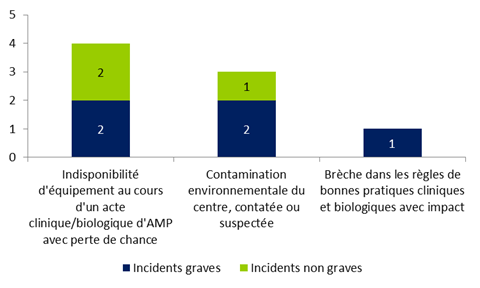
En 2015, 8 incidents relatifs à la structure ont été rapportés. Il s’agit pour plus de la moitié d’incidents graves (62,5%). La typologie la plus fréquente concerne une indisponibilité d'équipement au cours d'un acte clinique/biologique d'AMP avec perte de chance.
Erreurs d’identification
En 2015, 4 erreurs d’identification ont été déclarées et concernaient :
- une erreur d'attribution de spermatozoïdes découverte à la fin de l'insémination des ovocytes en fécondation in vitro classique. Les ovocytes n’ont pas été conservés ;
- une erreur d’attribution d’un embryon lors du transfert, liée à l’inattention du technicien et à l’organisation mise en place pour l’étiquetage des boîtes de transfert. La patiente a reçu un traitement abortif et le processus d’identitovigilance de l’établissement a été modifié ;
- une erreur d'étiquetage d’ovocytes au décours d’une ponction folliculaire aboutissant à la destruction du tube mal étiqueté. La procédure d’étiquetage du centre a été révisée suite à cet événement ;
- la mise en évidence d'une discordance sur l'identification de 2 paillettes d'embryons (les couleurs des joncs et des visiotubes utilisés différaient des informations transcrites dans le dossier patient, le registre et le système d’information). L’enquête n'a pas permis de lever le doute sur l'identification des embryons cryoconservés et les paillettes ont été détruites. La procédure de vitrification a été révisée suite à cet incident.
Ces erreurs d’identification ou d’attribution, dont l’une a abouti au transfert d’un embryon ne correspondant pas aux caractéristiques du couple, font parties des événements à impact fort justifiant, au-delà des mesures correctives prises localement, le renforcement des mesures barrières présentes dans les règles de bonnes pratiques en assistance médicale à la procréation et qui font actuellement l’objet d’une révision par l’Agence de la biomédecine.
Gravité / Conséquences
Tout comme pour les effets indésirables, la gestion des déclarations d’incidents par l’Agence de la biomédecine est notamment basée sur le niveau de gravité des conséquences des événements rapportés. Pour les incidents, seul le caractère grave ou non grave a été pris en compte.
L‘évolution des incidents en fonction de la gravité entre 2011 et 2015 est présentée dans le tableau TAMPV8.
Tableau TAMPV8. Evolution des incidents en fonction de la gravité
|
|
2011 |
2012 |
2013 |
2014 |
2015 |
|||||
N |
% |
N |
% |
N |
% |
N |
% |
N |
% |
|
Incidents non graves |
48 |
62,3 |
53 |
58,9 |
82 |
76,6 |
80 |
58,8 |
89 |
71,2 |
Incidents graves |
29 |
37,7 |
37 |
41,1 |
25 |
23,4 |
56 |
41,2 |
36 |
28,8 |
Total |
77 |
|
90 |
|
107 |
|
136 |
|
125 |
|
Les incidents non graves représentent plus de 70% des incidents rapportés et sont sans grande valeur ajoutée en matière de santé publique. Seuls 36 incidents graves ont été déclarés en 2015. Pour rappel, un incident est considéré comme grave s’il est susceptible d’entraîner des effets indésirables graves ou d’occasionner une erreur d’attribution ou une perte de gamètes, tissus germinaux ou embryons avec perte de chance totale de procréation sur la tentative ou pour le couple ou le patient.
Parmi les 125 incidents déclarés, 98 étaient susceptibles d’entraîner une perte de gamètes, embryons ou tissus germinaux et donc d’aboutir à une perte de chance qui a dans certains cas été évitée grâce à des mesures de récupération (mesures barrières). Ces incidents et leur impact sur les couples ou les patients sont présentés dans le tableau TAMPV9 ci-dessous.
Tableau TAMPV9. Impact des incidents sur les couples ou les patients
|
Gamètes |
Embryons |
Tissus germinaux |
Nombre d'incidents concernés |
31 |
64 |
3 |
Nombre de patients/ couples concernés |
55 |
189 |
478 |
Dont nombre de patients/couples avec une perte de gamètes, embryons ou tissus germinaux avérée ou potentielle |
|||
| - avec perte de procréation potentielle | 14 |
13 |
0 |
| - avec perte de procréation partielle | 8 |
27 |
0 |
| - avec perte de procréation totale | 5 |
18 |
0 |
Activité de don
Parmi les 125 incidents, seul 1 incident concernait le don de gamètes mais également les activités d’AMP et d’autoconservation. Il s’agissait de problèmes d'identité détecté dans plusieurs systèmes d’information d’un établissement mais sans erreur avérée grâce au contrôle effectué sur les dossiers papier. Cet événement s’est produit lors des fusions de dossiers entre le logiciel utilisé en AMP et celui utilisé dans l’établissement pour la gestion du dossier médical et le pilotage de la prise en charge en raison de divergences dans l’un ou l’autre des systèmes sur l’identité ou le numéro unique d’identification de certains patients (erreurs de saisie).
Activité d’autoconservation
Parmi les 14 déclarations d’incidents qui concernent l’autoconservation, 7 sont relatives à une autoconservation en vue de préservation de la fertilité et 7 à une autoconservation dans le cadre de l’AMP. La distribution de ces incidents en fonction de leur typologie ainsi que leur évolution entre 2011 et 2015 est représentée dans le tableau TAMPV10 ci-après.
Tableau TAMPV10. Evolution des incidents en autoconservation en fonction de leur typologie entre 2011 et 2015.
|
2011 |
2012 |
2013 |
2014 |
2015 |
|
Stimulation ovarienne |
Erreur ou anomalie d'administration |
1 |
0 |
0 |
0 |
0 |
Erreur d'attribution/identification |
Erreur d'identification/ d'attribution de gamètes ou d'embryons |
0 |
0 |
0 |
1 |
0 |
Culture et manipulation de gamètes/embryons |
Cultures contaminées (en dehors de la contamination liée à une infection de l'un ou l'autre des conjoints concernés) |
0 |
0 |
0 |
0 |
1 |
Perte de gamètes/embryons |
Maladresse ou incident lié à un environnement ou à un matériel particulier |
1 |
0 |
1 |
2 |
1 |
Conservation / décongélation inappropriée |
0 |
0 |
3 |
0 |
1 |
|
Transport inapproprié |
2 |
0 |
0 |
2 |
1 |
|
Equipement défectueux |
0 |
1 |
3 |
0 |
2 |
|
Perte de paillettes ou rupture de paillettes |
0 |
1 |
3 |
2 |
1 |
|
Perte de traçabilité |
0 |
0 |
1 |
0 |
0 |
|
Autres |
1 |
0 |
0 |
0 |
0 |
|
Total |
4 |
2 |
11 |
6 |
6 |
|
Structure (locaux, équipements, procédures) |
Perte de confidentialité ou de sécurité |
0 |
0 |
0 |
1 |
0 |
Indisponibilité d'équipement au cours d'un acte clinique/biologique d'AMP avec perte de chance |
0 |
1 |
0 |
0 |
0 |
|
Contamination environnementale du centre, constatée ou suspectée |
0 |
0 |
0 |
0 |
1 |
|
Brèche dans les règles de bonnes pratiques cliniques et biologiques avec impact |
0 |
2 |
0 |
0 |
0 |
|
Total |
0 |
3 |
0 |
1 |
1 |
|
Autres |
Non-respect des obligations réglementaires inhérent au couple |
0 |
0 |
1 |
0 |
0 |
Autres |
0 |
3 |
2 |
6 |
6 |
|
Total |
5 |
8 |
14 |
14 |
14 |
|
III. Répartition des déclarations par région
La répartition des événements indésirables en fonction de l’activité et des régions est représentée dans le tableau TAMPV11 ci-après
AMP |
Don |
Autoconservation |
||||
Région |
Nombre |
Fréquence |
Nombre |
Fréquence |
Nombre |
Fréquence |
Guadeloupe |
1 |
0,0% |
0 |
. |
0 |
. |
La Réunion |
6 |
83,3% |
0 |
. |
0 |
. |
Ile-de-France |
130 |
57,7% |
2 |
50 ,0% |
9 |
44,4% |
Champagne-Ardenne |
8 |
62,5% |
2 |
100% |
0 |
. |
Picardie |
22 |
81,8% |
0 |
. |
0 |
. |
Haute-Normandie |
14 |
85,7% |
0 |
. |
0 |
. |
Centre |
13 |
84,6% |
1 |
100% |
0 |
. |
Basse-Normandie |
9 |
66,7% |
1 |
100% |
0 |
. |
Bourgogne |
29 |
100% |
0 |
. |
0 |
. |
Nord - Pas-de-Calais |
15 |
80,0% |
0 |
. |
1 |
100% |
Lorraine |
5 |
100% |
0 |
. |
1 |
0,0% |
Alsace |
14 |
71,4% |
0 |
. |
0 |
. |
Franche-Comté |
9 |
100% |
0 |
. |
1 |
100% |
Pays de la Loire |
36 |
75,0% |
0 |
. |
2 |
50,0% |
Bretagne |
14 |
85,7% |
1 |
100% |
1 |
0,0% |
Poitou-Charentes |
1 |
100% |
0 |
. |
0 |
. |
Aquitaine |
22 |
86,4% |
0 |
. |
0 |
. |
Midi-Pyrénées |
9 |
88,9% |
0 |
. |
1 |
100% |
Limousin |
17 |
100% |
0 |
. |
0 |
. |
Rhône-Alpes |
31 |
80,6% |
0 |
. |
0 |
. |
Auvergne |
2 |
50,0% |
0 |
. |
0 |
. |
Languedoc-Roussillon |
21 |
85,7% |
0 |
. |
0 |
. |
Provence-Alpes-Côte d'Azur |
44 |
88,6% |
0 |
. |
1 |
0,0% |
En 2015, 83 centres d’AMP (centres clinico-biologiques d’AMP ou laboratoires d’IA) répartis dans 23 régions ont fait au moins une déclaration d’AMP vigilance. Dans 4 régions (Corse, Guyane, Martinique, Mayotte), les centres d’AMP n’ont fait aucune déclaration d’AMP vigilance. Dans les autres régions, en moyenne près de 21 déclarations d’AMP vigilance ont été faites par région avec des extrêmes allant de 1 déclaration à 130 déclarations.
Pour plus d’informations, le rapport annuel d’AMP vigilance 2015 adressé au ministre en charge de la santé est téléchargeable à partir de juillet 2015 sur le site de l’Agence de la biomédecine (http://www.agence-biomedecine.fr/professionnels/amp-vigilance.html)
1 Décret n°2008-588 du 19 juin 2008 transposant en matière de don de gamètes et d’assistance médicale à la procréation la directive 2004/23/CE du Parlement européen et du Conseil du 31 mars 2004
2 La date de déclaration correspond à l’envoi de la Partie A de la fiche de déclaration (déclaration immédiate)
3 Complications related to ovarian stimulation and oocyte retrieval in 4052 oocyte donor cycles. D. Bodri, J.J. Guillén, A. Polo et al – Reproductive BioMedicine Online, Vol. 17 n°2, 2008, 137-243.
The incidence of both serious and minor complications in young women undergoing oocyte donation. K.N. Maxwell, I.N.Cholst and Z. Rosenwaks – Fertility and Sterility, Vol. 90 n°6, 2008, 2165-2171.