Dispositif de biovigilance
La Biovigilance, créée en 20031, est organisée selon des modalités précisées par des décrets destinés à prendre notamment en compte le cadre européen des directives sur les tissus, les cellules et les organes2.
La biovigilance est une vigilance complexe portant à la fois sur des produits issus du corps humain et sur des activités allant de la sélection des donneurs au suivi post-greffe des receveurs.
Elle a la particularité de s’exercer sur des domaines médicaux hétérogènes présentant des risques variés en lien avec des activités thérapeutiques non systématiquement standardisées et en constante évolution.
Depuis 2003 et jusqu’au 30 novembre 2016, l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) était en charge du dispositif de la biovigilance. Elle a contribué à sa mise en place et à son organisation au sein du territoire français à travers le réseau des Correspondants Locaux de Biovigilance (CLB) et le développement d’outils méthodologiques comme le guide de biovigilance et la fiche de déclaration des événements indésirables. L’Agence de la biomédecine (ABM) a toujours été fortement impliquée dans ce dispositif puisque l’ensemble des déclarations devait lui être adressé en parallèle des déclarations faites à l’ANSM en raison de l’impact possible des événements indésirables sur les activités de régulation et de répartition des organes et des cellules.
Depuis le 1er décembre 2016 et en application de la loi de modernisation de notre système de santé, l’Agence de la biomédecine est désormais chargée d’assurer la mise en œuvre du dispositif de biovigilance. La « philosophie » initiale en a été modifiée avec la publication du décret n°2016-1206. En effet, à la déclaration sans délai de tous les événements indésirables succèdera progressivement un dispositif associant à la fois une surveillance par les professionnels de santé de la fréquence de survenue « effets indésirables attendus » (également dénommés « aléas thérapeutiques") et une déclaration sans délai des effets indésirables inattendus, des incidents graves et de toute fréquence anormalement élevée d’effets indésirables attendus ou d’incidents non graves.
Données générales 2016
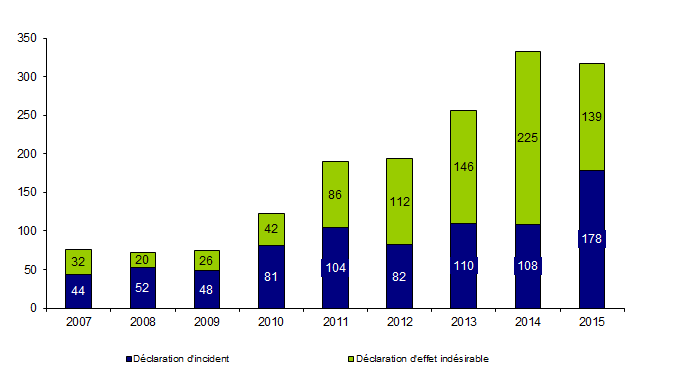
Evolutions du nombre de déclarations
Entre le 1er janvier 2016 et le 31 décembre 2016, l’Agence nationale de sécurité du médicament et des produits de santé puis l’agence de la biomédecine ont reçu 564 déclarations de biovigilance.
L’évolution du nombre de déclarations de biovigilance est présentée sur la figure BIOV1.
Figure BIOV1. Evolution du nombre de déclarations de biovigilance entre 2007 et 2016
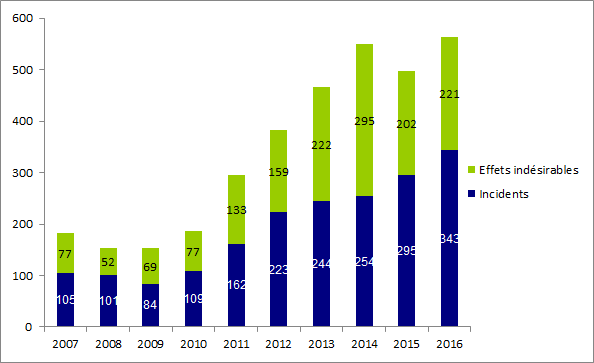
L’analyse du nombre de déclarations par année permet d’apprécier pour partie l’efficacité du dispositif mis en place. Le nombre de déclarations a pratiquement triplé depuis la mise en place du dispositif en 2008, néanmoins on observe une stabilisation du nombre de déclarations depuis 2014 (excepté en 2014 où le nombre de déclarations est supérieur d’environ 15 % par rapport aux autres années).
Effets indésirables et incidents
Les 564 déclarations de biovigilance sont réparties en 221 effets indésirables et en 343 incidents. Leur répartition en fonction des greffons concernés est représentée dans la figure BIOV1. Comme chaque année, la part des déclarations « organes » est majoritaire avec près de 63% du total, suivie par les déclarations « cellules » (31,2%), puis « tissus » (2,5%) et enfin « lait » (1,2%).
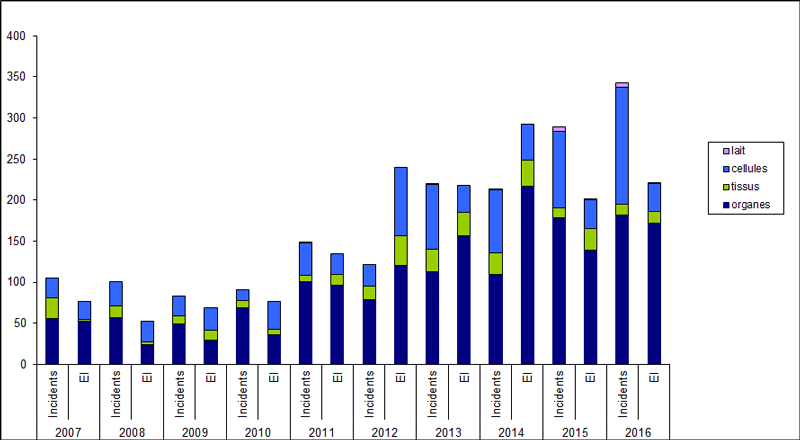
L’évolution au cours du temps de la répartition des déclarations entre effets indésirables et incidents et entre catégorie de greffons (organes, tissus, cellules et lait maternel) est représentée sur la figure BIOV3.
Figure BIOV2. Répartition des déclarations 2016 par catégorie de greffons
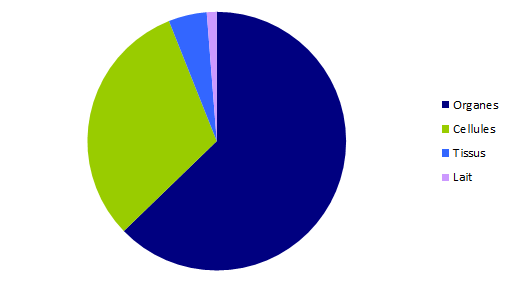
Figure BIOV3. Répartition des déclarations 2016 par catégorie de greffons et par type d’événements (incidents et effets indésirables EI)
Gravité des déclarations
La gestion des déclarations par l’Agence de la biomédecine est notamment basée sur le niveau de gravité des effets indésirables rapportés. Il existe 5 niveaux de gravité allant de G1 à G5. La figure BIOV4 présente la distribution des déclarations d’effets indésirables en fonction de leur gravité finale.
1-Négligeable : Manifestations cliniques ou biologiques ne nécessitant aucune prise en charge ou traitement médical.
2-Modérée : Manifestations cliniques ou biologiques sans menace vitale à court ou long terme et ne nécessitant pas d’hospitalisation.
3-Sévère : Manifestations cliniques ou biologiques entraînant une invalidité ou une incapacité, ou provoquant, prolongeant ou compliquant une hospitalisation ou tout autre état morbide, ou nécessitant une intervention médicale ou chirurgicale pour éviter un dommage permanent ou la défaillance d’une fonction corporelle.
A noter : les infections sévères susceptibles d’avoir été transmises par le produit biologique ou les activités de prélèvement ou de greffe/administration doivent systématiquement être déclarées et ceci à un niveau de gravité supérieur ou égal à 3.
4-Majeure : Menace vitale immédiate.
5-Décès.
Figure BIOV4. Distribution des déclarations en fonction de la gravité des effets indésirables
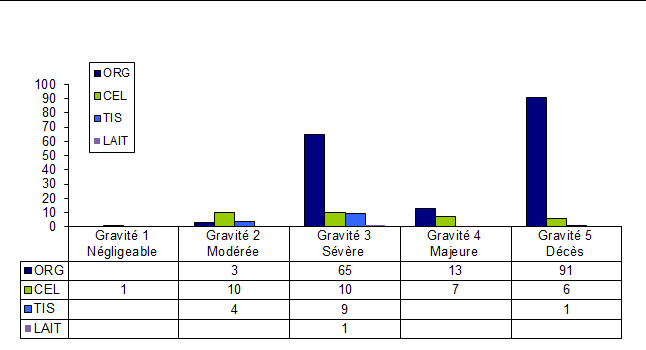
La très forte majorité des déclarations de biovigilance est de gravité G3 ou supérieure (92,1%) ce qui souligne la part importante des risques liés à ces activités thérapeutiques, ce qui doit être mis en regard avec les bénéfices majeurs attendus de ces actes de greffes.
Biovigilance Organes
Données générales
Le nombre total d’événements indésirables « organes » déclarés au cours de l’année 2016 est de 354 déclarations (soit 182 déclarations d’incidents et 172 déclarations d’effets indésirables parmi lesquelles 98,2% d’effets indésirables graves3).
Figure BIOV5. Nombre total d’événements indésirables « ORGANES » et évolution (2010-2016)
Les effets indésirables Organes
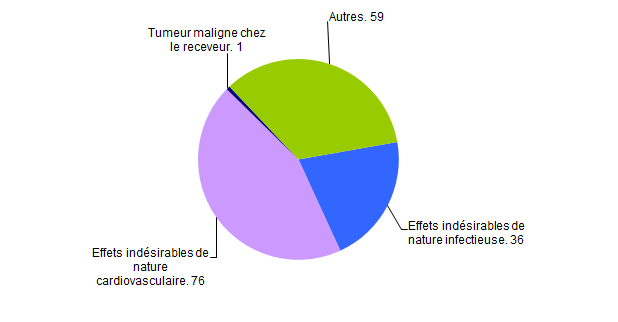
Parmi les 172 déclarations d’effets indésirables, on dénombre 76 déclarations d’effets indésirables de nature cardiovasculaire, 36 déclarations d’effets indésirables de nature infectieuse et 1 déclaration liée à une tumeur secondaire (métastases cérébrales chez un receveur cardiaque à 9 mois post-greffe). Leur répartition par type est représentée dans la figure BIOV6 ci-dessous.
Figure BIOV6. Déclaration d’effets indésirables de nature cardiovasculaire, infectieuse et tumorale (n=172, 2016)
|
Principales catégories d’effets indésirables au cours de l’année 2016 :
|
La répartition selon le délai de survenu de l’événement indésirable par rapport à l’acte de greffe des 91 déclarations d’effets indésirables ayant entrainé un décès du receveur est présentée dans l’histogramme BIOV7 ci-dessous.
Figure BIOV7. Répartition des déclarations concernant le décès d’un receveur en fonction du délai de survenue de l’effet indésirable par rapport à l’acte de greffe
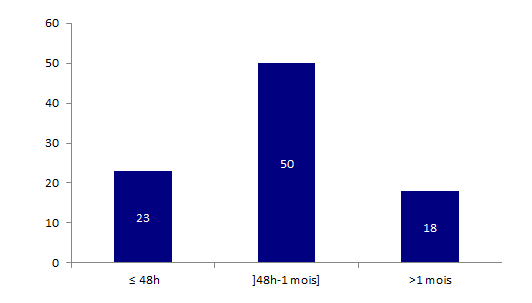
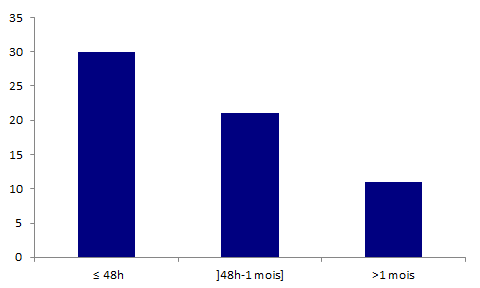
La répartition selon le délai de survenu de l’événement indésirable par rapport à l’acte de greffe des 62 déclarations d’effets indésirables ayant entrainé une détransplantation du receveur est présentée dans l’histogramme ci-dessous.
Figure BIOV8. Répartition des déclarations concernant des détransplantations en fonction du délai de survenue de l’effet indésirable par rapport à l’acte de greffe
Problématique rein (57)
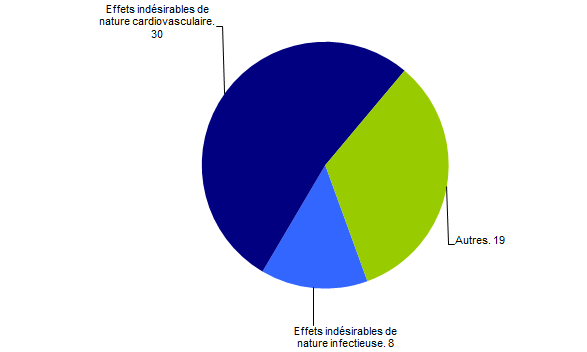
Concernant le rein, il convient de noter que parmi les 57 déclarations reçues en 2016, 30 étaient en lien avec une complication de nature cardiovasculaire et 8 liées à un événement infectieux. Le nombre de déclarations est représenté dans la figure BIOV9 ci-dessous.
Figure BIOV9. Problématique rein - déclaration d’effets indésirables de nature cardiovasculaire et infectieuse
Onze (11) déclarations d’effets indésirables ont entrainé le décès du receveur de rein :
Trente-sept (37) déclarations d’effets indésirables ont entrainé une détransplantation ou un arrêt fonctionnel du greffon (AFG).
Neuf (9) déclarations d’effets indésirables n’ont entrainé ni décès ni détransplantation chez le receveur.
Problématique pancréas (1)
Une (1) déclaration d’effet indésirable de nature cardiovasculaire (thrombose veineuse)
Problématique foie (41)
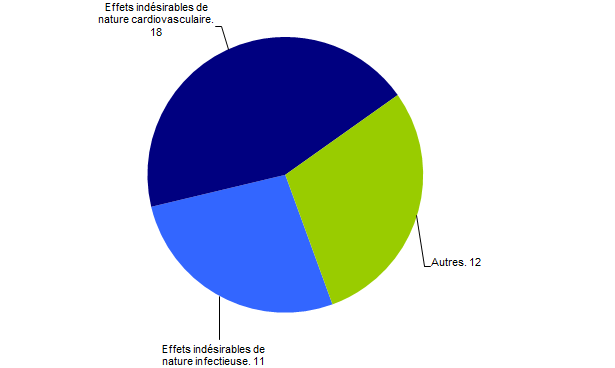
Concernant le foie, il convient de noter que parmi les 41 déclarations reçues en 2016, 18 étaient en lien avec une complication de nature cardiovasculaire et 11 liées à un événement infectieux. Le nombre de déclaration est représenté dans la figure BIOV10 ci-dessous.
Figure BIOV10. Problématique foie - déclaration d’effets indésirables de nature cardiovasculaire et infectieuse
24 déclarations d’effets indésirables ont entrainé un décès du receveur de foie
14 déclarations d’effets indésirables ont entrainé une détransplantation
Problématique cœur (36)
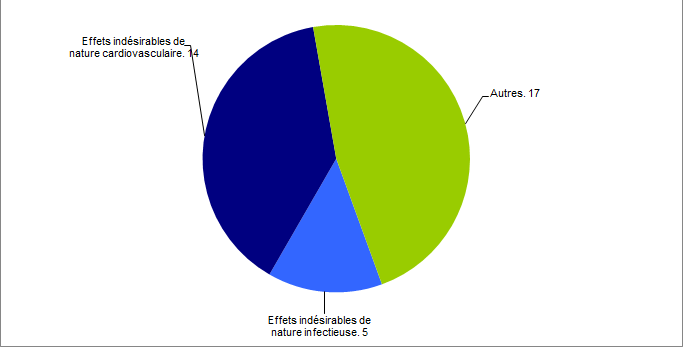
Concernant le cœur, il convient de noter que parmi les 36 déclarations reçues en 2016, 14 étaient en lien avec une complication de nature cardiovasculaire et 5 liées à un événement infectieux. Le nombre de déclarations est représenté dans la figure BIOV11 ci-dessous.
Figure BIOV11. Problématique cœur - déclaration d’effets indésirables de nature cardiovasculaire et infectieuse
Trente-trois (33) déclarations d’effets indésirables ont entrainé un décès du receveur de cœur :
Une (1) déclaration d’effet indésirable a entrainé une détransplantation (défaillance primaire du greffon cardiaque).
Une (1) déclaration d’effet indésirable était en lien avec la survenue d’une tumeur avec la découverte d’un cancer secondaire (métastases cérébrales) chez un receveur de cœur.
Problématique poumon (24)
Dix-neuf (19) déclarations d’effets indésirables ont entrainé un décès du receveur de poumon.
Une (1) déclaration d’effets indésirables a entrainé une détransplantation (rejet chronique)
Problématique des greffes combinées (13)
Greffes [Cœur-poumon] (4)
Deux (2) déclarations d’effets indésirables ont entrainé un décès du receveur de [cœur-poumon].
Une (1) déclaration d’effet indésirable a entrainé une détransplantation de poumon lors d’une greffe combinée [cœur-poumon]
Une (1) déclaration d’effet indésirable n’entrainant ni décès ni détransplantation chez le receveur a été déclarée pour une greffe combinée [cœur-poumon].
Greffes [poumon-foie] (1)
Une (1) déclaration de décès à J4 sur défaillance multiviscérale avec hémorragie, absence de contractibilité myocardique et insuffisance rénale.
Greffes [foie-rein] (4)
- Une (1) déclaration d’une réaction allergique à la noisette chez un receveur pédiatrique sans antécédent d’allergie connu avant greffe (recherche d’IgE anti noisette réalisée après ces manifestations allergiques positive chez le receveur et le donneur, le transfert d’allergie du donneur au receveur a été confirmé).
- Une (1) déclaration d’effet indésirable ayant entrainé un décès chez le receveur de foie-rein (séroconversion HSV1 chez le receveur et décès à J14 dans un contexte de syndrome respiratoire aigu, il est à noter une sérologie HSV1 négative chez le donneur mais une PCR HSV1 positive confirmant une primoinfection herpétique récente chez le donneur).
- Une (1) déclaration d’hémorragies diffuses (et dégradation fonctionnelle du foie) entrainant une détransplantation hépatique.
- Une (1) déclaration d’un arrêt fonctionnel du greffon rénal chez un receveur foie-rein.
Greffes [rein-pancréas] (4)
Quatre (4) déclarations d’effet indésirables de nature cardiovasculaire ont entrainé une détransplantation du pancréas (3 concernent des hémorragies, 1 concerne une thrombose artérielle et veineuse).
Les incidents Organes
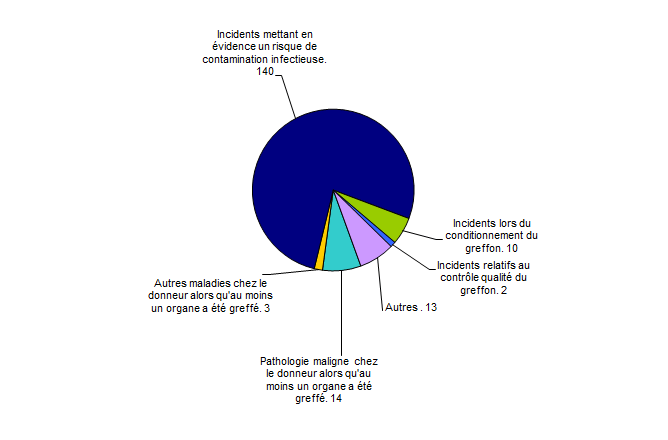
On dénombre 182 déclarations d’incidents « organes » dans la figure BIOV12 ci-dessous.
Figure BIOV12. Nombre de déclarations d'incidents par typologie (n=182, 2016)
Il convient de noter que parmi ces 182 déclarations, un nombre important concerne des événements porteurs de risque (ex : découverte de lésions suspectes au cours du processus de sélection d’un donneur / positivité d’un ECBU ou d’une hémoculture,…) qui ne sont pas la conséquence d’un accident ou d’une erreur et qui, par conséquence, ne répondent pas stricto sensu à la définition d’un incident de biovigilance.
Aussi, à compter du prochain rapport annuel de biovigilance (rapport 2017), les événements de cette nature ne seront plus comptabilisés et présentés dans le rapport. En effet, la réalisation de ces examens fait partie intégrante des procédures de sélection des donneurs et de contrôle de la qualité des greffons et à ce stade (c’est-à-dire en l’absence de tout effet indésirable en lien avec cet événement porteur de risque), il n’y a aucune mesure corrective à mettre en œuvre puisque celles-ci ont été respectées.
Répartition des déclarations d’incidents par étape de survenue dans le processus allant du prélèvement à la greffe et au suivi post greffe
Les événements porteurs de risque qui ne sont pas des incidents de biovigilance, ne sont pas comptabilisés dans ce chapitre. Aussi seules 163 déclarations d’incidents sur 182 sont présentées dans l’histogramme de répartition par étape de survenue présenté ci-après.
Figure BIOV13. Répartition des incidents par étape de survenue dans le processus
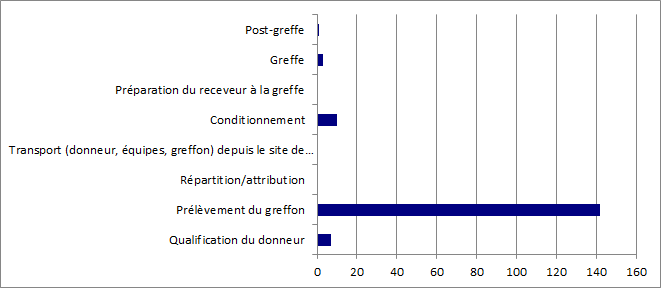
La répartition des déclarations d’incidents par étape de survenue dans le processus allant du prélèvement à la greffe et au suivi post greffe montre que les incidents déclarés surviennent principalement à l’étape de prélèvement du greffon. 97% (138/142) et concernent essentiellement des contaminations de liquides de conservation d’organe. Il convient de noter que par principe les contaminations de liquide de conservation ont été rattachées à l’étape du prélèvement bien qu’il puisse y avoir théoriquement des contaminations qui se produisent soit lors du déconditionnement du greffon à l’étape de la greffe, soit lors de l’échantillonnage du liquide de conservation pour contrôle microbiologique, soit encore lors de la réalisation des techniques microbiologiques de contrôle. Il est à noter que ces incidents sont le plus souvent sans conséquence clinique chez les receveurs
Biovigilance Cellules
Données générales
176 déclarations « cellules » (34 déclarations d’effets indésirables (EIs) et 142 déclarations d’incidents) ont été reçues en 2016 en biovigilance.
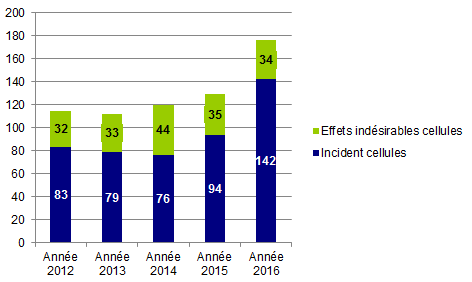
Ce chiffre est en nette augmentation par rapport aux années précédentes comme le montre l’histogramme BIOV14 ci-dessous.
Figure BIOV14. Répartition des déclarations « cellules » entre 2012 et 2016
Les effets indésirables Cellules
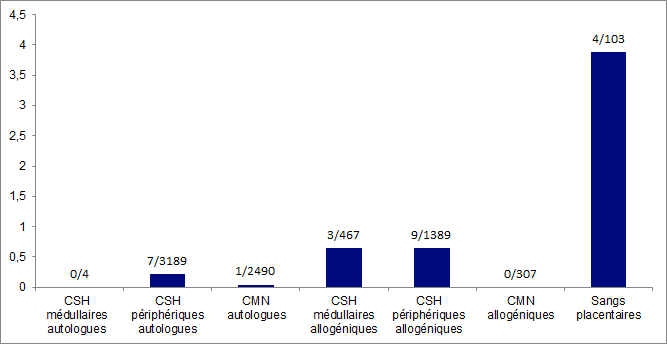
La figure ci-dessous illustre les taux d’EIs par type de greffons administrés rapportés au nombre d’actes de greffe réalisés.
Le nombre d’effets indésirables receveurs rapporté au nombre d’actes de greffe est assez faible compte tenu d’une part de la gravité des indications thérapeutique concernées et d’autre part de la lourdeur de leur prise en charge : 0.64EI pour les greffes de CSH médullaires, 0,65 EI pour les greffes de CSH périphériques, 0 pour les greffes de cellules mononucléées. Cela peut être le reflet d’une évaluation de ces évènements, survenus en post greffe, comme des aléas thérapeutiques attendus par les cliniciens en charge de ces patients et qui de ce fait ne les déclarent pas.
Dans le cadre de la mise en œuvre du nouveau décret sur la Biovigilance, l’Agence de la biomédecine va travailler avec les professionnels pour établir un référentiel de ces effets indésirables attendus considérés comme des aléas thérapeutiques des procédures de greffes de cellules souches hématopoïétiques. L’établissement de référentiels d’effets indésirables attendus a pour but de permettre aux professionnels d’identifier de façon plus ciblée les effets indésirables à surveiller et ceux qui le cas échéant seront à déclarer.
Figure BIOV15. Répartition des déclarations des EIs pour 100 administrations
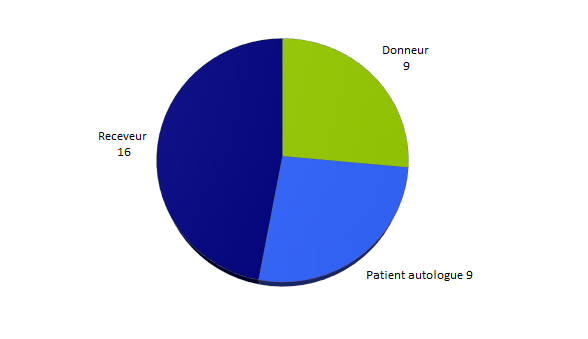
Les effets indésirables répertoriés concernent par ordre de fréquence, les receveurs de greffons allogéniques (16 déclarations), puis à égalité les donneurs (9) et les patients ayant reçus un greffon autologue (9).
Figure BIOV16. Répartition des déclarations d’effets indésirables par personnes concernées
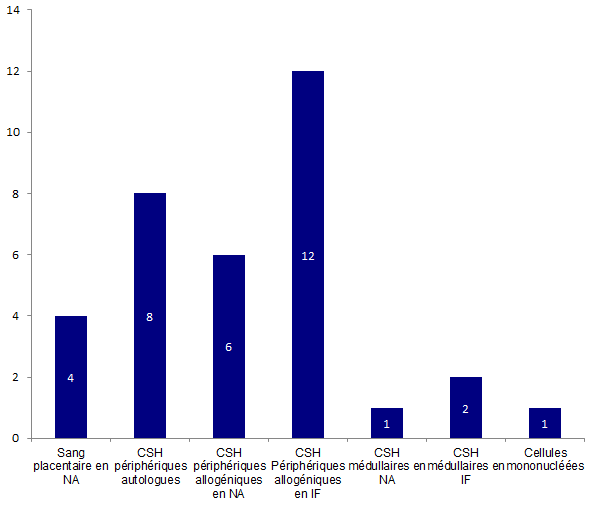
Les effets indésirables sont plus fréquemment constatés lors des prélèvements ou greffes de CSH périphériques d’origine intrafamiliale (IF) ou non apparenté (NA), (18 déclarations). Huit (8) déclarations concernent des effets indésirables survenus chez des patients ayant reçus des greffons autologues de CSH périphériques.
Seules 3 déclarations d’EIs concernent les CSH médullaires que ce soit pour des cellules d’origines intrafamiliales (2) ou non-apparentées (1).
Enfin, 4 déclarations relèvent de la greffe de sang placentaire non-apparenté et seulement une déclaration concerne les cellules mononucléées (CMN).
Figure BIOV17. Nombre de déclarations d'effet indésirable par type de greffon
Les EIs ont été typés pour être regroupés en grandes catégories en utilisant la terminologie MedDRA (le choix de cette terminologie médicale standardisée pour le typage des effets indésirables en biovigilance est actuellement en test à l’Agence de la biomédecine. Ce choix est basé sur la richesse de cette classification, par ailleurs déjà utilisée dans de nombreux domaines médicaux et notamment d’autres dispositifs de vigilances comme la pharmacovigilance). Deux niveaux de classifications ont été retenus, un niveau élevé ou général, les SOC (System Organ Class) et un niveau bas intermédiaire plus précis, les PT (Preferred Term).
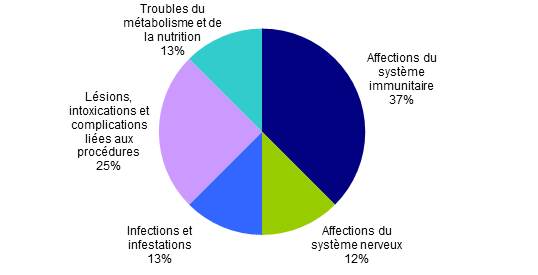
Concernant les effets indésirables liés au prélèvement ou à l’utilisation des CSH et CMN, on constate que trois catégories majeures peuvent être identifiées : 11 déclarations relèvent de la catégorie des « Affections du système immunitaires » ; 6 déclarations des « Lésions ; intoxications et complications liés aux procédures » et enfin 4 déclarations des « Infections et infestations ». Les autres déclarations se répartissent ensuite dans 9 autres catégories de la terminologie MedDRA qui en propose au total 27. Le tableau ci-dessous présente la répartition des EIs par SOC et PT.
Tableau BIOV1. Effets indésirables cellules par typologie MedDRA
|
Type d'effets indésirables MedDRA |
Nombre de déclarations |
SOC |
Affections cardiaques |
1 |
PT |
Péricardite |
1 |
SOC |
Affections du système immunitaire |
11 |
PT |
Allo-immunisation |
1 |
Choc anaphylactique |
2 |
|
Maladie du greffon contre l'hôte dans le tractus gastro-intestinal |
1 |
|
Réaction post-greffe |
5 |
|
Rejet de greffe |
1 |
|
Urticaire |
1 |
|
SOC |
Affections du système nerveux |
2 |
PT |
Scotome |
1 |
Syndrome d'encéphalopathie postérieure réversible |
1 |
|
SOC |
Affections hématologiques et du système lymphatique |
2 |
PT |
Microangiopathie thrombotique |
1 |
Syndrome myélodysplasique |
1 |
|
SOC |
Infections et infestations |
4 |
PT |
Bactériémie |
1 |
Choc septique |
2 |
|
Pneumonie |
1 |
|
SOC |
Lésions, intoxications et complications liées aux procédures |
6 |
PT |
Echec de prise de greffe |
4 |
Prise de greffe retardée |
2 |
|
SOC |
Troubles du métabolisme et de la nutrition |
3 |
PT |
Deshydratation |
1 |
Hypocalcémie |
2 |
|
SOC |
Troubles généraux et anomalies au site d'administration |
3 |
PT |
Asthénie |
2 |
Malaise |
1 |
|
SOC |
Tumeurs bénignes, malignes et non précisées (incl kystes et polypes) |
2 |
PT |
Angiomyolipome rénal |
1 |
Cancer du rein |
1 |
|
|
Total général |
34 |
Les effets indésirables des « CSH périphériques allogéniques »
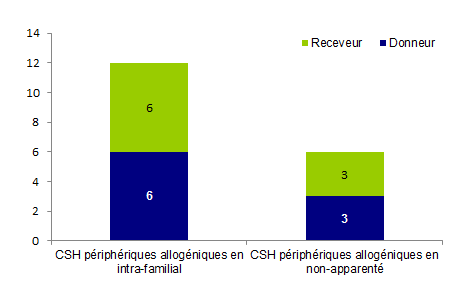
Le nombre de déclarations d’EIs concernant les CSH périphériques allogéniques est de 18. Elles se répartissent à égalité entre donneurs (9 déclarations) et receveurs (9 déclarations).On constate qu’un plus grand nombre de ces déclarations concernent les dons intrafamiliaux tant du côté des donneurs que des receveurs comme le montre l’histogramme BIOV18 ci-dessous.
Figure BIOV18. Répartition des EIs (CSH périphériques allogéniques) donneur/receveur
Les effets indésirables des « CSH médullaires allogéniques »
Seulement 3 déclarations d’effets indésirables concernant des CSH médullaires allogéniques ont été reçues en 2016, ce qui est en baisse par rapport aux années précédentes. L’utilisation des greffons de CSH médullaires qui avait été en forte baisse s’est désormais stabilisée et il est probable qu’elle augmente à nouveau avec le recours de plus en plus fréquent aux greffes haplo-identiques pour lesquelles certaines équipes préfèrent greffer des CSH médullaires
Les effets indésirables des « CSH périphériques autologues »
Le nombre de déclarations concernant des effets indésirables liés au prélèvement ou à l’administration de CSH périphériques autologues reste peu élevé avec 8 déclarations. Les effets indésirables peuvent être constatés au moment du prélèvement et au moment de la greffe et il convient de noter qu’une seule de ces déclarations concerne l’étape du prélèvement. Les effets indésirables se répartissent dans les catégories suivantes :
Figure BIOV19. Répartition des effets indésirables (SOC) des patients ayant reçus un greffon autologue
Les effets indésirables des « CSH d’origine placentaire »
Le nombre de déclarations d’EIs en greffe de sang placentaire reste stable et bas ; on compte 4 déclarations reçues en 2016. Il a été déclaré 2 chocs anaphylactiques survenus dans les premiers temps de la greffe, un rejet de greffe et un échec de prise de greffe.
Les effets indésirables des « cellules mononucléées»
Il n’y a eu en 2016 qu’une seule déclaration d’effet indésirable concernant les cellules mononucléées. Cette déclaration concerne des cellules mononucléées autologues puisque l’évènement est survenu au décours d’un traitement par photochimiothérapie extracorporelle. La patiente a présenté une bactériémie d’évolution favorable sous traitement.
Les effets indésirables des « Autres cellules »
Il n’y a eu aucune déclaration d’effet indésirable impliquant d’autres cellules que les cellules souches hématopoïétiques ou les cellules mononucléées.
Les incidents Cellules
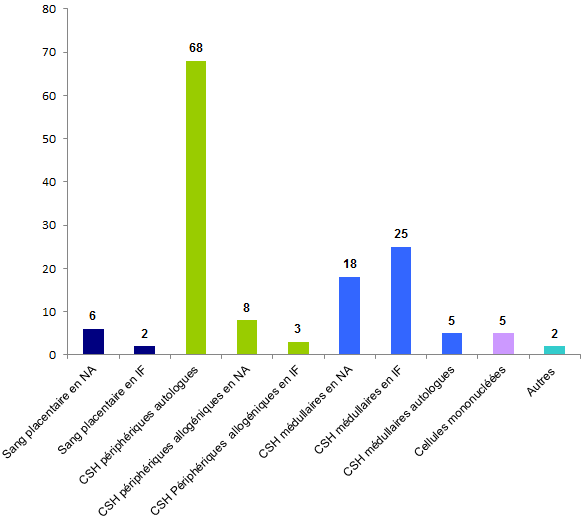
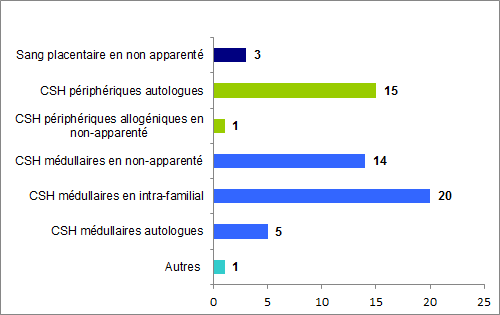
En 2016, 142 déclarations d’incidents ont été reçues dont la plus grande majorité concerne les greffons constitués de CSH périphériques autologues, soit 68 déclarations, et les déclarations concernant les greffons constitués de CSH médullaires autologues et allogéniques, intrafamilial ou non apparenté, représentent 48 déclarations.
Figure BIOV20. Répartition des déclarations "Incident" par type de greffons
On constate que les incidents surviennent essentiellement à l’étape de prélèvement et à l’étape de préparation du greffon, totalisant ainsi 130 déclarations.
Figure BIOV21. Nombre d’incidents par étape de survenue
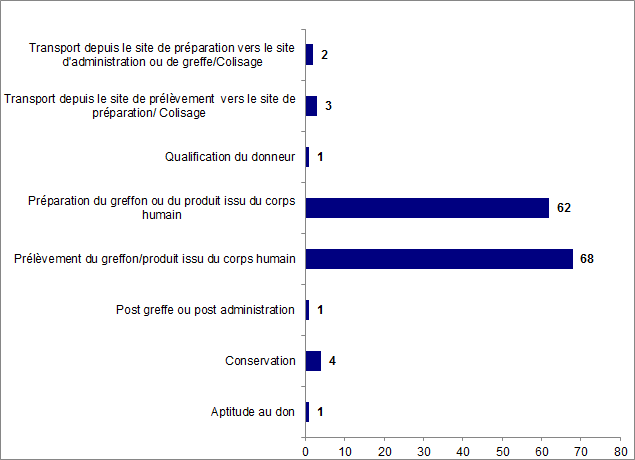
En 2016, les 142 déclarations d’incidents survenus au cours de la chaine thérapeutique allant du prélèvement à l’administration des produits de thérapie cellulaire ont principalement concerné :
- Des contrôles bactériologiques positifs avec des résultats obtenus en post-administration essentiellement liés à des germes commensaux de la flore cutanée et qui sont retrouvés principalement sur les greffons médullaires et les greffons de CSP autologues.
Figure BIOV22. Nombre de contaminations bactériennes par type de greffons
- Des incidents faisant part de greffons non conformes (quantité cellulaire insuffisante du greffon, mauvais rendement après décongélation en cellules CD34+ ou en CD3+ (pour les cellules mononucléées), hématocrite du greffon trop élevée ) pouvant entrainer ou entrainant un retard de prise en charge ou une fois greffé, une sortie d’aplasie tardive.
Au sein de cette catégorie, le nombre important de déclarations du type « mauvais rendement en cellules CD34+ » sera le point de départ d’une réflexion à l’Agence de la biomédecine par le biais d’un groupe de travail. Cette réflexion a déjà été initiée dans certaines unités de thérapie cellulaire sans que ne se soit dégagée de cause évidente expliquant ces mauvais rendements et donc rendant difficile l’élaboration de nouvelles procédures en vue de l’amélioration des pratiques.
- Des problématiques liées au conditionnement des greffons lors de leur prélèvement, transformation ou décongélation. Ces déclarations ont été adressées en parallèle en matériovigilance dès lors qu’un dispositif médical était potentiellement impliqué.
- Des problématiques liées aux transports des greffons.
- Des problématiques liées à la conservation des greffons : il a notamment été déclaré un incident en relation avec les cuves d’azote utilisées pour la conservation de greffons. La remontée en température accidentelle de ces cuves a entrainé la décongélation d’un nombre important de greffons, soient 226 poches pour 89 patients. Cet incident s’est produit suite à une cascade d’erreurs humaines à la fois lors du remplissage des tankers extérieurs (oubli d’ouverture de vanne) et lors du déclenchement des alarmes (non-respect des procédures). Une inspection de l’ANSM a alors été programmée. Un incident du même type ayant été déclaré en 2015 ainsi que deux incidents d’AMPvigilance impliquant des cuves de conservation, il a été décidé la création d’un groupe de travail sur la sécurisation du contenu des récipients cryogéniques
Biovigilance Tissus
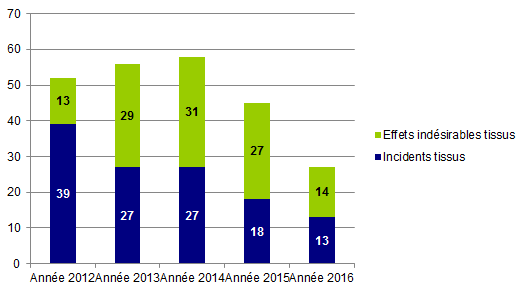
Le nombre de déclarations d’évènements indésirables concernant les greffes de tissus reste particulièrement faible au regard du nombre de produits greffés et a été en 2016 en baisse par rapport aux années précédentes comme le montre la figure ci-dessous. Les données chiffrées des années précédentes concernaient également des événements observés lors d’essais cliniques ce qui peut pour partie expliquer cette différence quantitative (ces essais étant aujourd’hui terminés). A noter que le nouveau dispositif de biovigilance ne couvre plus la vigilance des essais cliniques réalisés avec des produits issus du corps humains. La mise en œuvre de cette vigilance relève de la responsabilité de l’ANSM en charge également d’autoriser ces protocoles d’essais cliniques.
Cette faible activité en vigilance peut témoigner à la fois d’une relative bonne sécurité des greffons tissulaires mais également d’une sous notification probablement liée à la méconnaissance par les professionnels des exigences en matière de vigilance ou comme pour les organe et les cellules d’une non déclaration des effets indésirables attendus dans le cadre de la prise en charge des patients. La formation des CLB et des professionnels est une des actions cibles de l’Agence de la biomédecine (cf chapitre IV.8) et une première version devrait pouvoir être mise à disposition des professionnels à partir de 2018.
Figure BIOV23. Evolution des déclarations tissus depuis 2012
La majorité des déclarations concerne les greffes de cornée (14 déclarations) ; le reste des déclarations reçues se répartissant ensuite à peu près également entre les autres types de tissus.
Figure BIOV24. Répartition des déclarations par type de tissus
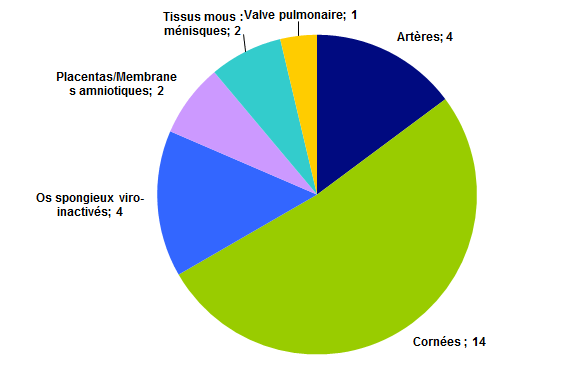
Figure BIOV25. Répartition effet indésirable/incident par type de tissus
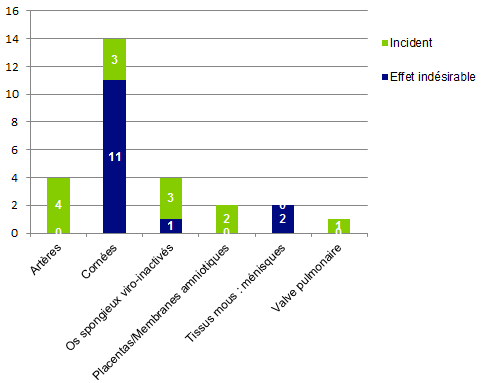
Les événements liés aux « Cornées »
Parmi les 14 déclarations cornées, 11 sont le fait d’un effet indésirable et 3 sont le fait d’un incident.
Dans le tableau ci-dessous, on retrouve le type des effets indésirables « cornées » regroupés en grandes catégories en utilisant comme pour les effets indésirables cellules, la terminologie MedDRA, par SOC (System Organ Class) et par PT (Preferred Term).
Tableau BIOV2. Effets indésirables tissus par typologie MedDRA
Type d’effets indésirables Med DRA |
Effets indésirables Cornée |
||
SOC |
Affections oculaires |
5 |
|
PT |
|
Œdème de la cornée |
5 |
SOC |
Infections et infestations |
1 |
|
PT |
|
Endophtalmie |
1 |
SOC |
Lésions, intoxications et complications liées aux procédures |
5 |
|
PT |
|
Complication lors d'une intervention |
1 |
|
Echec de prise de greffe |
4 |
|
|
|
Total |
11 |
Neuf de ces déclarations « cornée » relèvent d’un signalement groupé émanant d’un seul centre participant à une étude de suivi mise en place en 2013 (voir le rapport annuel de biovigilance de l’ANSM 2013, chapitre 4.2) et dont les conclusions seront établies par l’ANSM. Aucune de ces déclarations n’évoque de problématique liée à la qualité du greffon cornéen.
Le cas d’endophtalmie recensé est d’origine infectieuse et le germe retrouvé est un Streptococcus mitis/oralis. Les contrôles microbiologiques des milieux de cornée réalisés à la banque et au bloc opératoire lors de la greffe ont été retrouvés stériles.
Les 3 incidents « cornées » ont concerné : une contamination bactérienne et deux erreurs organisationnelles ou humaines lors du transport du greffon.
Les événements liés aux « Vaisseaux et valves »
Quatre déclarations concernant des greffes d’artères ont été adressées à la cellule de biovigilance ; toutes concernent des incidents du type « contaminations bactériennes ». Pour chacun de ces cas, le liquide de décongélation a présenté des contrôles bactériologiques positifs. Aucun des patients greffés n’a présenté de symptômes particuliers par la suite. Il s’agissait le plus souvent de germes appartenant à la flore cutanée (Staphylococcus warneri, Propionibacterium avidum ou acnes).
Il n’a été déclaré qu’un seul évènement en relation avec une greffe valvulaire ; il s’agit d’un incident du type « contamination bactérienne » du liquide de conservation.
Les événements « Autres tissus »
Os viro inactivés
Quatre déclarations ont été reçues, dont une en rapport avec un effet indésirable et trois avec des incidents.
- L’effet indésirable déclaré est de gravité importante puisque le patient est décédé dans les suites d’un choc septique ; les germes Enterobacter cloacae et Streptococcus agalactiae ont été retrouvés dans le sang et dans l’exsudat du péricarde. La greffe a eu lieu en Espagne et les tissus osseux ont été préparés (2 têtes fémorales) par une banque française pour laquelle l’ANSM a décidé a posteriori d’organiser une inspection. L’enquête n’a pas retrouvé d’élément relatif à un défaut de la stérilisation permettant d’identifier une cause liée aux greffons.
- 3 incidents ont été déclarés et qui sont des dysfonctionnements de la gestion et de la traçabilité des tissus. Ces incidents n’ont pas eu de retentissement clinique sur les patients Des mesures correctives ont été mises en place.
Placentas/Membranes amniotiques
Deux déclarations relatives à un incident ont été reçues : une erreur de conditionnement du greffon et une erreur d’attribution. Ces incident bien qu’enregistrés comme incident de biovigilance relèvent plutôt du management de la qualité ; après enquête, des mesures correctives ont été prises.
Tissus mous/Ménisques
Deux déclarations d’effets indésirables ont été reçues : des paresthésies et des douleurs post opératoires qui ont nécessité une exploration diagnostique arthroscopique.
Biovigilance Lait maternel
Seules 7 déclarations d’incidents et d’effets indésirables en lien avec les activités liées au lait maternel à usage thérapeutique ont été adressées en biovigilance pendant l’année 2016. Ce nombre reste particulièrement faible.
Parmi les 7 déclarations reçues, on retrouve 6 incidents et un effet indésirable.
Trois déclarations d’incidents survenus dans le cadre de l’utilisation du Lait pasteurisé issu de dons personnalisés.
Deux déclarations d’incidents survenus dans le cadre de l’utilisation du Lait pasteurisé issu de dons anonymes.
Une déclaration d’incident survenu dans le cadre de l’utilisation du lait maternel cru issu de dons personnalisés.
Une déclaration d’un effet indésirable :
Cas groupés d’entérocolites retrouvés dans le service de néonatologie. Des prélèvements systématiques ont retrouvés la présence de Bacillus dans l’environnement du lactarium. Les investigations ont révélé la non-conformité des désinfectants fournis avec un nouveau pasteurisateur. Une décision de suspension provisoire d’activité au sein du lactarium a été mise en œuvre le temps nécessaire à la réalisation d’une mise à blanc de l’environnement et d’une désinfection conforme des pasteurisateurs.
1 Décret n°2003-1206 du 12 décembre 2003, décret n° 2007-1110 du 17 juillet 2007 relatif à la biovigilance et à l’hémovigilance, décret 2016-1622 du 29 novembre 2016 relatif aux dispositifs de biovigilance et d’AMP vigilance.
2 Directive 2006/86/CE du 24 octobre 2006 de la Commission portant application de la directive 2004/23/CE du Parlement européen et du Conseil en ce qui concerne les exigences de traçabilité, la notification des réactions et incidents indésirables graves, (…) des tissus et cellules d’origine humaine et la directive 2005/45/UE du Parlement européen et du Conseil du 7 juillet 2010 relative aux normes de qualité et de sécurité des organes humains destinés à la transplantation.
3 : effet indésirable susceptible d'entraîner la mort ou de mettre la vie en danger, d'entraîner une invalidité ou une incapacité, ou de provoquer ou de prolonger une hospitalisation ou tout autre état morbide, ou susceptible de se reproduire chez un ou plusieurs patients, donneurs vivants ou receveurs