Dispositif de biovigilance
La surveillance de la sécurité et de la qualité de l’acte de greffe ou d’administration d’un produit issu du corps humain est une préoccupation constante pour les professionnels concernés par ces activités et pour les autorités de santé et tout particulièrement l’Agence de la biomédecine. Cette surveillance s’exerce avec des techniques variées depuis les premières données pré-cliniques sur le modèle animal jusqu’à la mise en place d’essais cliniques ou de protocoles sur l’homme. Ces recherches comportent, le plus souvent, une évaluation de l’efficacité de l’acte thérapeutique bien plus développée que les objectifs de sécurité ne serait-ce qu’en raison des contraintes liées à l’effectif minimal de l’échantillon de receveurs pour pouvoir observer des différences de risque significatives. Qu’il s’agisse d’essais cliniques, de protocoles ou de procédures, les professionnels de santé et les autorités compétentes tendent à la minimisation des risques par la mise en place de mesures de prévention (ex : prévention des risques liés au geste clinique par la délivrance d’autorisation de sites ou d’habilitation des professionnels, prévention des risques liés aux voyages du donneur par la mise en œuvre de recommandations/exigences concernant leur éviction au don ou leur dépistage, etc.).
Ces données de sécurité « a priori » étant le plus souvent parcellaires, la surveillance de l’équilibre bénéfices/risques, quel que soit le produit prélevé, greffé ou administré, repose essentiellement sur les actions d’évaluation pouvant être menées dans le cadre d’un usage dit « de routine », c’est-à-dire auprès d’une population beaucoup plus large et variable.
Cette surveillance « a posteriori » constitue le fondement de la biovigilance qui repose aussi bien sur l’analyse des déclarations des professionnels de santé réceptionnées au fil de l’eau que sur les données épidémiologiques issues de la surveillance de populations cibles (ex : donneurs de CSH, patients greffés rénaux, nourrissons receveurs de lait maternel issu de lactarium…). L’apport des données épidémiologiques, à l’instar de la pharmaco-épidémiologie, palie à la fois le risque de sous-notification, la difficulté d’identification des signaux faibles (ceux pour lesquels l’imputabilité du greffon ou de l’acte de greffe n’est pas certaine) et l’absence de possibilité de quantification du risque (liée pour partie à la sous-notification).
C’est cette « bio-surveillance » dont la mise en place se fera progressivement qui constitue le nouveau cadre d’une biovigilance modernisée ayant pour objet la détection, l’évaluation et la quantification des risques liés à l’usage des produits issus du corps humain en tenant compte de toutes les données exploitables : caractéristiques du greffon, indications du prélèvement, de la greffe ou de l’administration du produit, données des essais cliniques, déclarations spontanées, études épidémiologiques, données de la littérature scientifique, analyses des bases de données médico-administratives.
L’année 2017 a essentiellement été consacrée à l’élaboration de la méthodologie permettant de déterminer quels sont les événements indésirables attendus et acceptables au regard du bénéfice de l’acte thérapeutique étudié. Ce travail devra être poursuivi en 2018 avec l’aide de groupes de travail composés par des représentants des sociétés savantes et permettre à terme de publier des référentiels de risques dans chaque domaine (organes, tissus, cellules, lait). Ces référentiels permettront aux professionnels de santé de distinguer parmi l’ensemble de ces événements, ceux qui devront faire l’objet d’une déclaration immédiate, d’une surveillance active, d’un recueil annuel ou d’une surveillance passive.
Cette clarification du champ permettra de prendre en considération la part de risque inhérente aux activités de prélèvement, de greffes ou d’administrations des éléments ou produits issus du corps humain couvertes par la biovigilance tout en conservant la réactivité nécessaire à la mise en place de mesures correctives lors de l’observation d’événements inacceptables en matière de santé publique ou au regard de la prise en charge individuelle du donneur ou du patient concerné.
Données générales 2017
Evolution du nombre de déclarations
Figure BIOV1. Evolution du nombre de déclarations
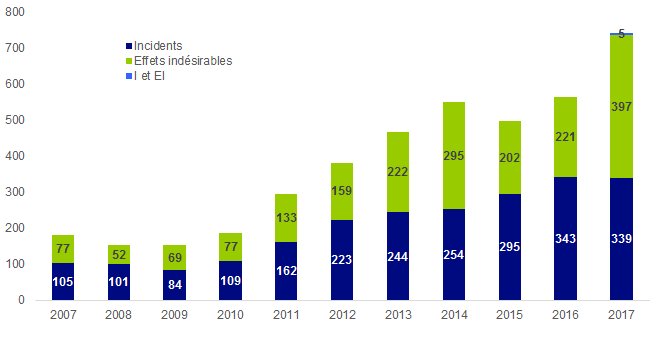
Entre le 1er janvier 2017 et le 31 décembre 2017, l’Agence de la biomédecine a reçu 741 déclarations de biovigilance.
L’évolution du nombre de déclarations de biovigilance est présentée sur la figure BIOV1.
Les 741 déclarations de biovigilance sont réparties en 339 incidents, 397 effets indésirables et 5 incidents et effets indésirables liés.
Effets indésirables et incidents
Figure BIOV2. Répartition des déclarations par type de greffons
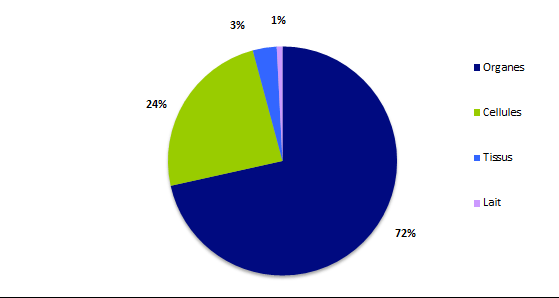
Comme chaque année, la part des déclarations « organes » est majoritaire avec près de 72% du total suivie par les déclarations « cellules » (24%) puis « tissus » (3%) et enfin « lait » (1%).
Figure BIOV3. Evolution de la répartition des déclarations depuis 2007 par catégorie de greffons et d'évènements
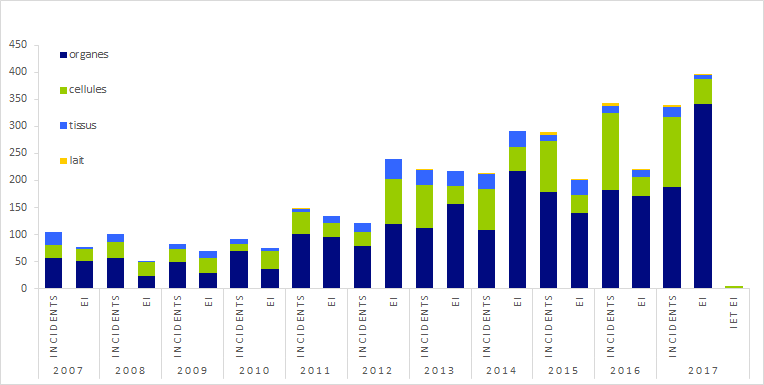
L’évolution au cours du temps de la répartition des déclarations entre effets indésirables et incidents et entre catégories de greffons (organes, cellules, tissus, lait) est représentée sur la figure BIOV3.
Gravité des déclarations
La gestion des déclarations par l’Agence de la biomédecine est notamment basée sur le niveau de gravité des effets indésirables rapportés. Il existe 5 niveaux de gravité allant de G1 à G5. La figure BIOV4 présente la distribution des déclarations d’effets indésirables en fonction de leur gravité finale.
1-Négligeable : Manifestations cliniques ou biologiques ne nécessitant aucune prise en charge ou traitement médical.
2-Modérée : Manifestations cliniques ou biologiques sans menace vitale à court ou long terme et ne nécessitant pas d’hospitalisation.
3-Sévère : Manifestations cliniques ou biologiques entraînant une invalidité ou une incapacité, ou provoquant, prolongeant ou compliquant une hospitalisation ou tout autre état morbide, ou nécessitant une intervention médicale ou chirurgicale pour éviter un dommage permanent ou la défaillance d’une fonction corporelle.
A noter : les infections sévères susceptibles d’avoir été transmises par le produit biologique ou les activités de prélèvement ou de greffe/administration doivent systématiquement être déclarées et ceci à un niveau de gravité supérieur ou égal à 3.
4-Majeure : Menace vitale immédiate.
5-Décès.
Figure BIOV4. Distribution des déclarations en fonction de la gravité des effets indésirables
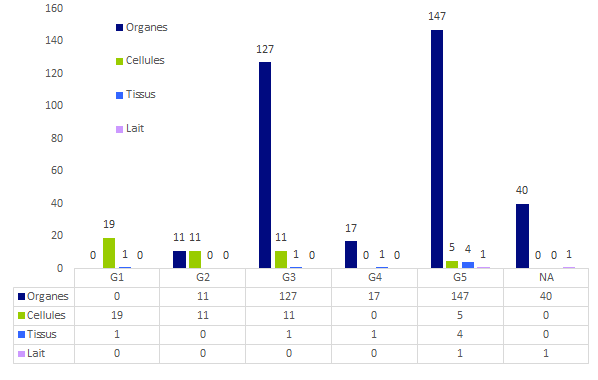
*NA : non applicable du fait de données manquantes
Une très forte majorité des déclarations présente au moins une gravité supérieure ou égale à G3 ce qui souligne la part importante des risques liés aux activités thérapeutiques des domaines envisagés.
Les évènements de gravité G1 et G2 sont une part importante des déclarations « cellules » et sont surtout le fait des EI donneurs et dans une moindre part des EI concernant les patients autologues.
Biovigilance Organes
Données générales
Figure BIOV5. Evolution des déclarations de biovigilances organes depuis 2007
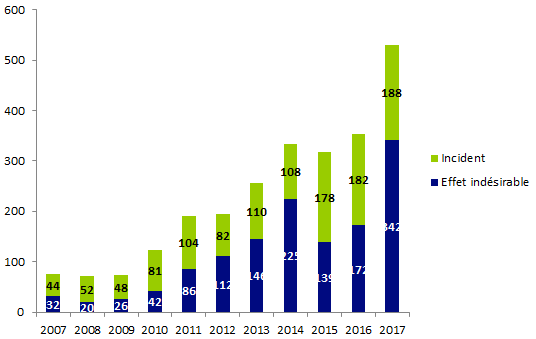
Le nombre total d’évènements indésirables « organes » déclarés au cours de l’année 2017 est de 530 déclarations (soient 188 déclarations incidents et 342 déclarations d’effets indésirables).
Depuis le nouveau décret de biovigilance du 29 novembre 2016 et le transfert de cette mission à l’Agence de la biomédecine, la politique d’incitation à la déclaration par les CLB des décès et des détransplantations dont l’Agence avait connaissance a été remplacée par un dispositif de déclarations directes aux CLB via le Pôle sécurité-qualité afin que celui-ci précise les caractéristiques de l’événement indésirable et apporte des informations concernant les mesures correctives mises en place le cas échéant. Cette nouvelle attitude explique en partie l’augmentation importante des déclarations « effets indésirables » observée en 2017.
Chiffres généraux EI organes
Les effets indésirables répartis selon l’organe impliqué sont représentés dans la figure BIOV6 ci-après.
Figure BIOV6. Effets indésirables « organes » par type de greffons
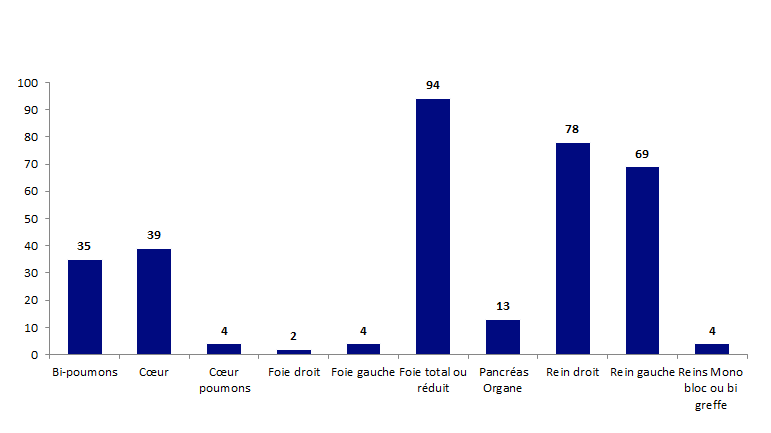
Figure BIOV7. Répartition des EI déclarés par nombre de greffes
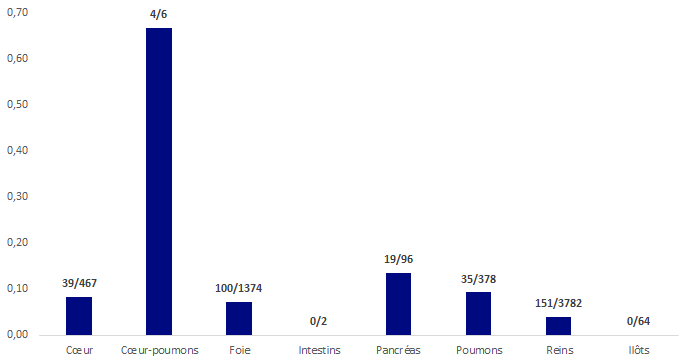
L’histogramme ci-dessus illustre les taux d’EI receveurs par type de greffons greffés/administrés - toutes gravités et niveaux d’imputabilité confondus. Le taux global des notifications d’effets indésirables receveurs rapporté au nombre total d’administrations/greffes réalisées est de 0,005% (soit 1 effet indésirable toutes les 2000 greffes).
Chiffres généraux incidents organes
Figure BIOV8. Répartition des incidents par étape de survenue
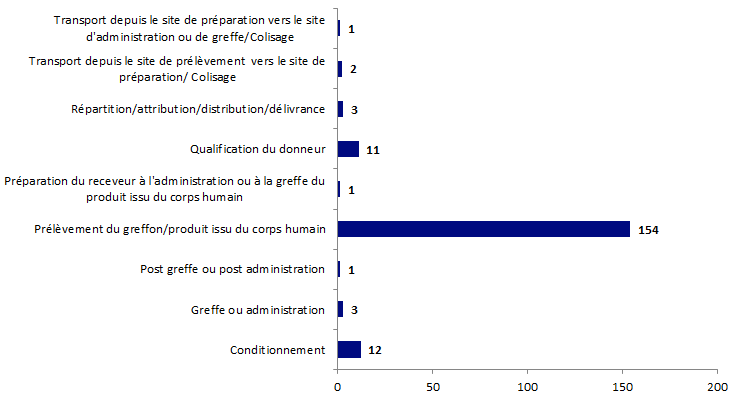
En 2017, 188 déclarations incident organes ont été reportées. Ces 188 déclarations concernent essentiellement les greffons rénaux (102 déclarations) et les greffons hépatiques (52 déclarations).
Comme les années précédentes, l’Agence de la biomédecine de par ses activités opérationnelles est le principal déclarant des incidents survenant sur la chaine allant du prélèvement à la réception du greffon notamment des contaminations des liquides de transport à agents fongiques.
Pour plus d’informations et de détails sur les déclarations d’effets indésirables et d’incidents - organes, le rapport annuel de Biovigilance 2017 est téléchargeable à partir de juillet 2018 sur le site de l’agence de la biomédecine : https://www.agence-biomedecine.fr/Biovigilance-organes
Biovigilance cellules
Les déclarations cellules
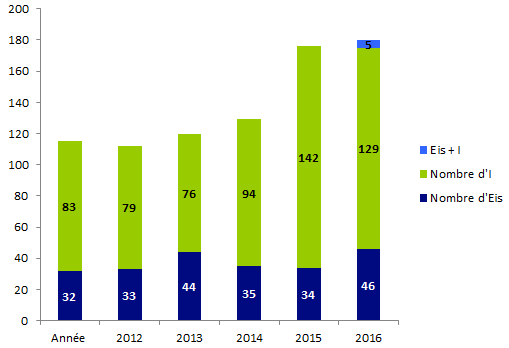
180 déclarations de biovigilance « cellules » (46 déclarations d’effets indésirables, 129 déclarations d’incidents et 5 déclarations d’incidents et d’effets indésirables liés) ont été rapportées en 2017 à l’Agence de la biomédecine.
Ce chiffre est stable par rapport à l’année précédente comme le montre la figure ci-dessous.
Ces déclarations ne concernent que des cellules d’origine hématopoïétique : soit des cellules souches d’origine médullaire (CSH médullaires) ou périphérique (CSP), soit des cellules mononucléées (CMN).
Aucun autre type de cellules n’a fait l’objet d’une déclaration.
Figure BIOV9. Répartition des déclarations cellules entre 2012 et 2017
Les effets indésirables (EI) cellules
Figure BIOV10. Répartition des EI déclarés rapportés au nombre de greffes (receveurs autologues et allogéniques)
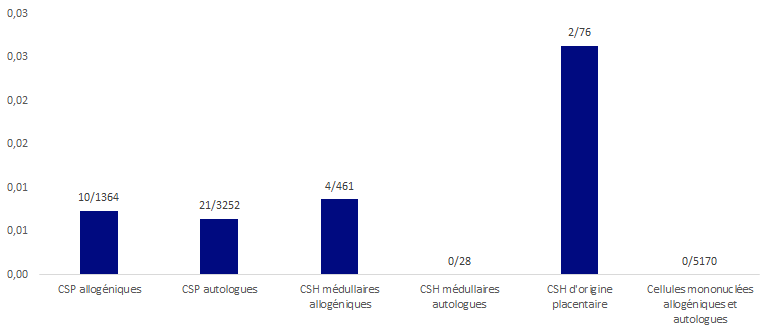
L’histogramme ci-dessus illustre les taux d’EI receveurs par type de greffons greffés, toutes gravités et niveaux d’imputabilité confondus. Le taux global de notification d’effets indésirables receveurs rapporté au nombre total d’administrations/greffes réalisées est de 0,003% (soit 1 effet indésirable toutes les 3000 greffes).
Figure BIOV11. Répartition des EI par personne concernée
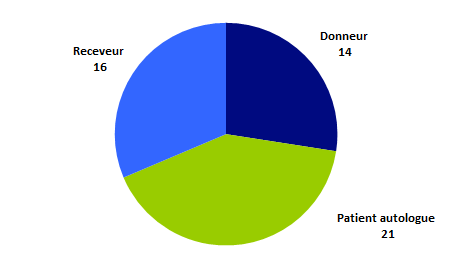
Les effets indésirables répertoriés concernent par ordre de fréquence : les patients autologues (21 déclarations) puis les receveurs allogéniques (16 déclarations) et enfin les donneurs allogéniques (14 déclarations dont 9 déclarations en intrafamilial et 5 en non-apparenté).
Les incidents (I) cellules
Chiffres généraux incidents cellules
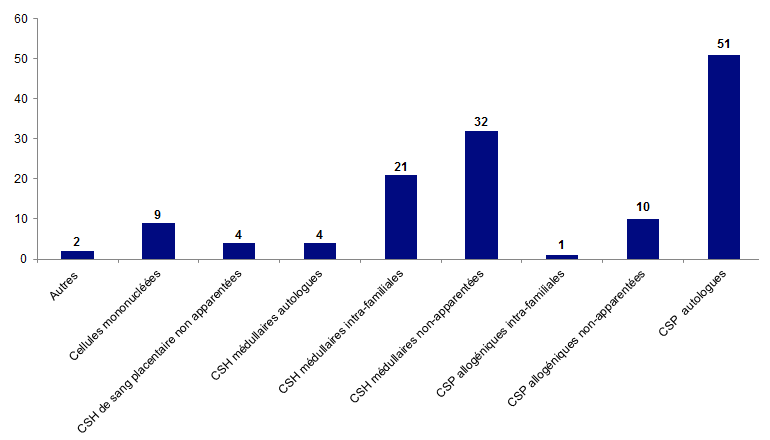
En 2017, 134 déclarations d’incidents ont été reçues dont la plus grande majorité concerne les greffons de CSH autologues issues du sang périphérique, soient 51 déclarations.
La catégorie « autres » représentée sur les différentes figures du chapitre incident ne concerne pas d’autres cellules que les CSH ou les CMN, elle est le fait de déclarations impliquant plusieurs types de produits cellulaires à la fois ; par exemple lors d’incidents impliquant les cuves de conservation de greffons ou lors de problématiques plus globale sur la qualification du greffon des CSH médullaires autologues et allogéniques, des CSH périphériques autologues et allogéniques, …
Figure BIOV12. Répartition des déclarations cellules incident par type de greffon
Le typage des incidents
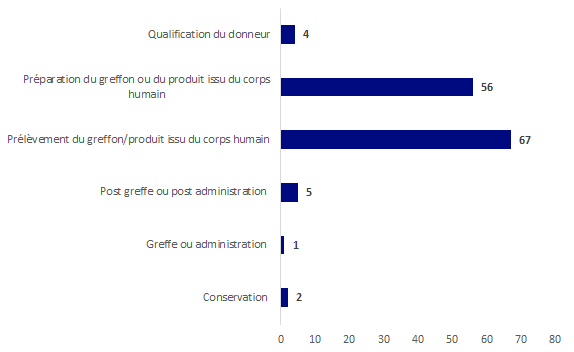
Les incidents ont été typés par étapes : ils surviennent essentiellement au prélèvement et à la préparation comme illustré sur la figure ci-dessous.
Figure BIOV13. Répartition par étape des déclarations incident cellules
Pour plus d’informations et de détails sur les déclarations d’effets indésirables et d’incidents - cellules, le rapport annuel de Biovigilance 2017 est téléchargeable à partir de juillet 2018 sur le site de l’agence de la biomédecine : https://www.agence-biomedecine.fr/biovigilance-cellules
Biovigilance Tissus
Les déclarations tissus
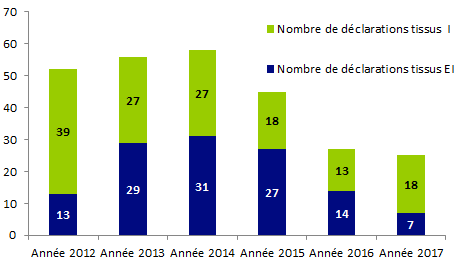
Le nombre de déclarations d’évènements indésirables concernant les tissus reste particulièrement faible (25 déclarations) au regard du nombre de produits greffés ; par ailleurs, en 2017 ce nombre est en discrète baisse par rapport aux années précédentes.
Figure BIOV14. Evolution des déclarations « tissus » depuis 2012
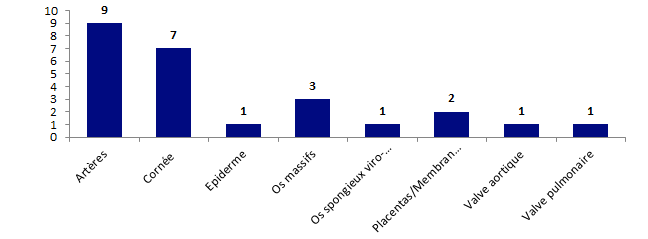
En 2017, les déclarations concernent essentiellement les greffes de cornée (7 déclarations) et les greffes d’artères (9 déclarations) comme le montre la figure ci-dessous.
Figure BIOV15. Répartition des déclarations par type de tissus
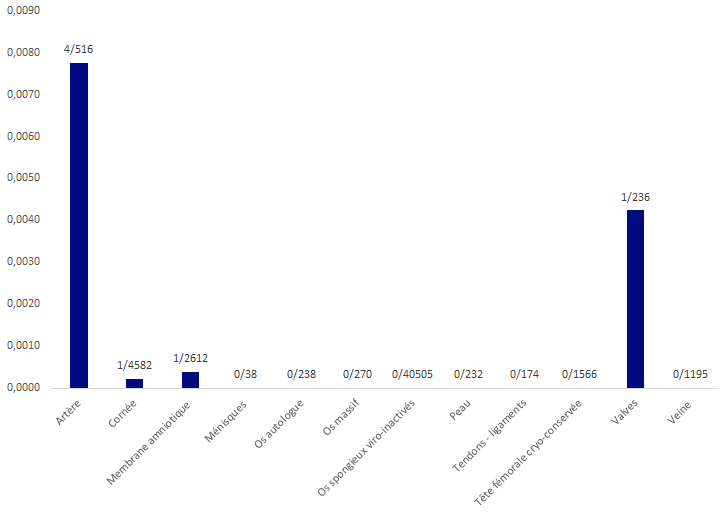
La figure ci-dessous illustre les taux d’EI receveurs par type de greffons greffés.
Ce taux reste bas et préjuge vraisemblablement d’une sous notification.
Figure BIOV16. Répartition des EI déclarés rapportés au nombre de greffes (receveurs autologues et allogéniques)
Pour plus d’informations et de détails sur les déclarations d’effets indésirables et d’incidents - tissus, le rapport annuel de Biovigilance 2017 est téléchargeable à partir de juillet 2018 sur le site de l’agence de la biomédecine: https://www.agence-biomedecine.fr/Biovigilance-Tissus
Biovigilance Lait
Les déclarations Lait
Seules 6 déclarations d’incidents et d’effets indésirables en lien avec les activités liées au lait maternel à usage thérapeutique ont été adressées en biovigilance durant l’année 2017. Il s’agit d’un nombre restant faible au regard des activités de délivrance de lait maternel sur le territoire national.
Il est fort probable que le dispositif de biovigilance dans ce domaine ne reflète pas les incidents et les effets indésirables observés sur le terrain : ce constat peut être le reflet d’une méconnaissance du dispositif de biovigilance par les professionnels impliqués malgré les échanges réguliers avec l’Agence de la biomédecine.
Pour plus d’informations et de détails sur les déclarations d’effets indésirables et d’incidents - lait, le rapport annuel de Biovigilance 2017 est téléchargeable à partir de juillet 2018 sur le site de l’agence de la biomédecine : https://www.agence-biomedecine.fr/Biovigilance-Lait-Maternel
Actions 2017
Mise en place des comités de vigilance (COVI)
Suite au transfert de la biovigilance à l’Agence de la biomédecine, il a été décidé de mettre en place des comités de vigilance (COVI) pour aider l’Agence dans ses missions d’évaluation et dans ses choix stratégiques pour les domaines suivants : organes, tissus, cellules et lait.
Lors de ces COVI, il est prévu l’analyse de certaines déclarations complexes, la mise en place d’un programme de travail (priorisation des référentiels de risques), la préparation d’enquêtes ou d’études, la diffusion, le cas échéant des recommandations en découlant, la proposition de thématiques de minimisation de risques, déterminée soit selon les déclarations de biovigilance reçues à l’Agence, soit selon les publications récentes.
Mise en place de l’outil de télédéclaration BioVigie
En 2017, L’Agence de la biomédecine a poursuivi avec un prestataire informatique le développement du système de télédéclarations BIOVigie afin de permettre aux correspondants locaux de biovigilance (CLB) d’effectuer des déclarations et d’y renseigner les mesures correctives mises en œuvre. Il permettra aussi les échanges entre l’Agence et les CLB lors des investigations et la gestion de leurs déclarations.
Cette application concerne tous les domaines de la biovigilance, c’est-à-dire les organes, les tissus, les cellules, les préparations de thérapie cellulaire et le lait.
Donneurs de CSH et risques thromboemboliques
En 2015, une déclaration faisait état de la survenue d’un accident thromboembolique grave diagnostiqué chez une donneuse de cellules souches hématopoïétiques (CSH) dans la semaine suivant son don de CSH périphériques.
Suite à cet événement, un groupe de travail a été mis en place. Il a finalement été conclu que la coexistence de plusieurs facteurs de risques chez la donneuse avait contribué à la survenue de l’embolie pulmonaire. Cependant, un facteur de risque a été identifié tout particulièrement car évitable : la pose d’un cathéter veineux fémoral pour la cytaphérèse. La solution de minimisation des risques proposée après cet événement est la modification du texte des Bonnes Pratiques de prélèvement, afin de restreindre cet acte aux seuls cas où il est impossible de changer de source cellulaire du fait d’une contre-indication au don de moelle et lorsque le prélèvement par voie veineuse périphérique s’avère irréalisable.
Maitrise de la phase pré analytique des échantillons de qualification des donneurs
En 2017, un groupe de travail associant des représentants des sociétés savantes (Société française de microbiologie et Société française de biologie clinique), l’établissement français du sang, l’Agence nationale de sécurité du médicament et des produits de santé a été constitué pour rédiger une mise au point sur la maitrise de la phase pré-analytique des échantillons destinés à la qualification microbiologique des donneurs (d’organes, de tissus et de cellules). Cette thématique avait été initiée lors de la réception d’une déclaration de biovigilance faisant état d’un résultat faussement positif pour le VIH lors de la qualification d’un donneur (sans impact pour les receveurs). Cette mise au point est consultable sur le site internet de l’Agence (https://www.agence-biomedecine.fr/Maitrise-preanalytique).
Mise en place d’un groupe de travail référentiel lait maternel à usage thérapeutique
L’Agence de la biomédecine a mis en place un groupe de travail « référentiel lait maternel à usage thérapeutique » avec la participation de l’association des lactariums de France (ADLF). Le but de ce groupe de travail est de publier un outil d’aide à la déclaration des incidents concernant le lait maternel préparé par les lactariums.
Pour plus d’informations et de détails, le rapport annuel de Biovigilance 2017 est téléchargeable à partir de juillet 2018 sur le site de l’agence de la biomédecine :
https://www.agence-biomedecine.fr/Biovigilance-organes
https://www.agence-biomedecine.fr/biovigilance-cellules
https://www.agence-biomedecine.fr/Biovigilance-Tissus
https://www.agence-biomedecine.fr/Biovigilance-Lait-Maternel