AMP Vigilance
- Qu’est-ce que l’AMP vigilance ?
- Les acteurs de l’AMP vigilance
- La déclaration d’AMP vigilance
- Le rapport annuel du CLA
- Le guide pratique d’aide à la mise en place de l’AMP vigilance dans un établissement de santé
- Les rapports annuels d’AMP vigilance
- Les réunions des correspondants locaux d’AMP vigilance
- La lettre d’information « AMP vigilance’ infos »
- Les mises au point / Recommandations
- Les textes juridiques
- Vos interlocuteurs à l’Agence de la biomédecine
Qu’est-ce que l’AMP vigilance ?
L’AMP vigilance consiste à surveiller, hors « essais cliniques » [1], les risques liés à l’utilisation à des fins thérapeutiques des gamètes, embryons et tissus germinaux dans le cadre d’activités cliniques et biologiques d’assistance médicale à la procréation (AMP) et à effectuer des déclarations ciblées (cf. infra). Cette surveillance des événements indésirables s’accomplit à chaque étape du processus : recueil, prélèvement, préparation, conservation, transport, mise à disposition, importation, exportation, greffe, insémination, transfert et étape post-insémination ou post-transfert.
L’AMP vigilance repose sur :
![]() la surveillance de façon systématique de tous les incidents et de tous les effets indésirables ;
la surveillance de façon systématique de tous les incidents et de tous les effets indésirables ;
![]() le signalement sans délai, par l’ensemble de ces professionnels, de tout incident grave et de tout effet indésirable inattendu au correspondant local d’AMP vigilance (CLA) ;
le signalement sans délai, par l’ensemble de ces professionnels, de tout incident grave et de tout effet indésirable inattendu au correspondant local d’AMP vigilance (CLA) ;
![]() la déclaration par le CLA sans délai des incidents graves et des effets indésirables inattendus à l’Agence de la biomédecine ;
la déclaration par le CLA sans délai des incidents graves et des effets indésirables inattendus à l’Agence de la biomédecine ;
![]() l’analyse et l’évaluation locale et nationale de ces informations en vue de limiter la probabilité de survenue de tout nouvel incident grave ou effet indésirable inattendu ou d’en diminuer la gravité.
l’analyse et l’évaluation locale et nationale de ces informations en vue de limiter la probabilité de survenue de tout nouvel incident grave ou effet indésirable inattendu ou d’en diminuer la gravité.
On entend par surveillance le fait pour les professionnels intervenants dans les activités d’AMP d’enregistrer tous les incidents et effets indésirables afin de signaler les incidents graves et les effets indésirables inattendus et de déterminer la fréquence de survenue des effets indésirables attendus. Les effets indésirables considérés comme attendus feront l’objet d’une publication par l’ABM sous forme de référentiels pour chaque indication. Les effets indésirables non répertoriés dans le référentiel seront considérés comme des effets indésirables inattendus et devront faire l’objet d’une déclaration immédiate à l’ABM
Incident grave :
![]() tout accident ou erreur entraînant ou susceptible d’entraîner un effet indésirable grave ou un effet indésirable inattendu, une erreur d’attribution des gamètes, tissus germinaux ou embryons, ou une perte importante de ces éléments du corps humain au cours de la tentative de procréation médicalement assistée ;
tout accident ou erreur entraînant ou susceptible d’entraîner un effet indésirable grave ou un effet indésirable inattendu, une erreur d’attribution des gamètes, tissus germinaux ou embryons, ou une perte importante de ces éléments du corps humain au cours de la tentative de procréation médicalement assistée ;
![]() toute fréquence anormalement élevée de survenue d’incidents ou d’effets indésirables attendus ;
toute fréquence anormalement élevée de survenue d’incidents ou d’effets indésirables attendus ;
![]() toute information concernant le donneur ou le don, découverte de façon fortuite après le prélèvement et dont les conséquences sont susceptibles d’entraîner un risque pour la santé des personnes qui ont recours à un don dans le cadre de l’assistance médicale à la procréation ou en sont issues ;
toute information concernant le donneur ou le don, découverte de façon fortuite après le prélèvement et dont les conséquences sont susceptibles d’entraîner un risque pour la santé des personnes qui ont recours à un don dans le cadre de l’assistance médicale à la procréation ou en sont issues ;
Effet indésirable : toute réaction nocive survenant chez les personnes prises en charge en AMP ou en étant issues.
Effet indésirable grave : tout effet indésirable ayant entraîné la mort ou ayant mis la vie en danger, entraîné une invalidité ou une incapacité, ou provoqué ou prolongé une hospitalisation ou tout autre état morbide ;
Effet indésirable inattendu (ou « inacceptable »), tout effet indésirable, grave ou non grave, dont la nature, la sévérité, l’évolution n’est pas attendue au regard des référentiels établis par l’Agence de la biomédecine ou compte tenu de l’état de santé de la personne ;
Effet indésirable attendu (ou « acceptable »), tout effet indésirable dont le risque de survenue est connu et mentionné dans le référentiel publié par l’Agence de la biomédecine dans l’indication thérapeutique concernée.
Les acteurs de l’AMP Vigilance
Les professionnels de santé
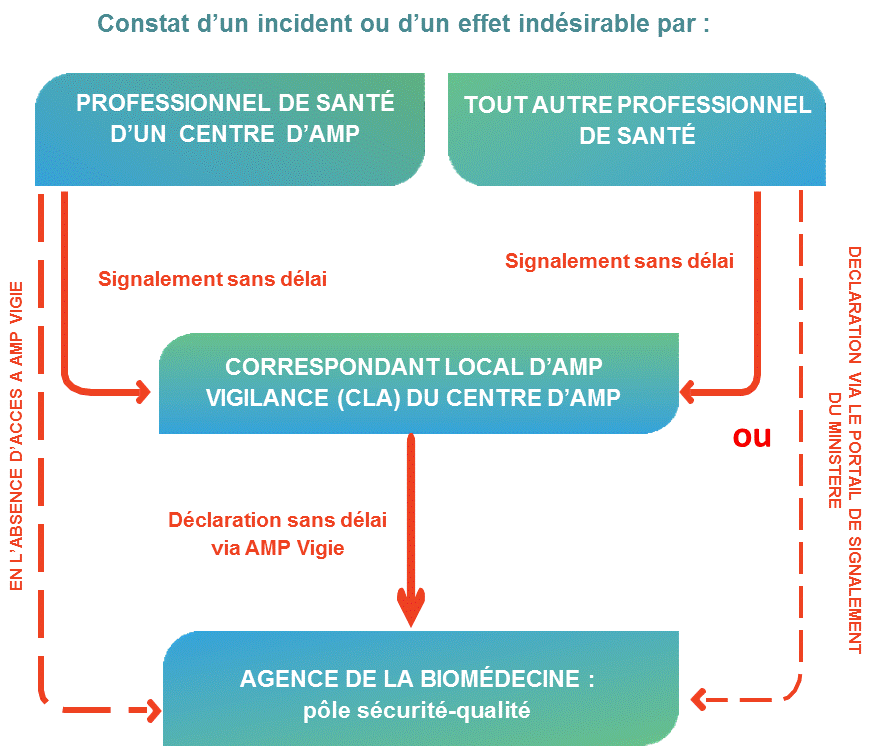
Les professionnels de santé qui participent à l’activité d’AMP, qu’ils pratiquent en établissement de santé ou dans un cadre libéral, jouent un rôle fondamental dans le système national d’AMP vigilance. Ce sont eux qui, lorsqu’ils ont connaissance d’un événement indésirable, ont pour mission d’en surveiller la fréquence de survenue et de signaler les incidents graves et les effets indésirables inattendus.
Le plus souvent, ces signalements sont adressés au correspondant local d’AMP vigilance de l’établissement qui en fera la déclaration à l’Agence de la biomédecine via l’outil de télédéclaration « AMP Vigie ».
En l’absence de correspondant local (cas essentiellement des professionnels exerçant dans un cadre libéral), le professionnel peut effectuer sa déclaration à l’Agence de la biomédecine en passant par le portail de signalement du ministère (https://signalement.social-sante.gouv.fr) ou en se mettant en relation avec le centre d’AMP ayant pris en charge le couple ou le patient afin que ce dernier fasse la déclaration auprès de l’Agence de la biomédecine.
L’Agence de la biomédecine (ABM)
L’Agence de la biomédecine est l’autorité compétente en matière d’AMP vigilance, son rôle consiste notamment à :
![]() évaluer toutes les déclarations d’incidents ou d’effets indésirables qui lui parviennent. Pour cela, elle s’appuie sur une expertise interne et le cas échéant peut faire appel à des experts externes. Elle est également en lien avec les autres vigilances concernées (matériovigilance, pharmacovigilance,..) qui ont comme autorité compétente l’ANSM.
évaluer toutes les déclarations d’incidents ou d’effets indésirables qui lui parviennent. Pour cela, elle s’appuie sur une expertise interne et le cas échéant peut faire appel à des experts externes. Elle est également en lien avec les autres vigilances concernées (matériovigilance, pharmacovigilance,..) qui ont comme autorité compétente l’ANSM.
![]() répondre aux demandes de tout interlocuteur en matière d’AMP vigilance ;
répondre aux demandes de tout interlocuteur en matière d’AMP vigilance ;
![]() animer le réseau en allant à la rencontre des acteurs sur le terrain ou en organisant une réunion nationale des CLA ;
animer le réseau en allant à la rencontre des acteurs sur le terrain ou en organisant une réunion nationale des CLA ;
![]() informer les professionnels de santé des mises au point et recommandations établies suite aux déclarations d’AMP Vigilance reçues ;
informer les professionnels de santé des mises au point et recommandations établies suite aux déclarations d’AMP Vigilance reçues ;
![]() mettre en place et animer des groupes de réflexions scientifiques et méthodologiques ;
mettre en place et animer des groupes de réflexions scientifiques et méthodologiques ;
![]() former les différents acteurs du dispositif d’AMP vigilance ;
former les différents acteurs du dispositif d’AMP vigilance ;
![]() analyser les rapports de synthèse d’AMP vigilance des CLA et rédiger, en partie à partir de ces rapports, le rapport annuel d’AMP vigilance destiné au ministre de la santé et à la Commission européenne.
analyser les rapports de synthèse d’AMP vigilance des CLA et rédiger, en partie à partir de ces rapports, le rapport annuel d’AMP vigilance destiné au ministre de la santé et à la Commission européenne.
Les correspondants locaux d’AMP vigilance (CLA)
Coordinateur : la section 3 du code de la santé publique (Art. R. 2142-19 à R. 2142-20) prévoit que, dans le centre d’AMP les membres de l’équipe médicale clinico-biologique et dans les laboratoires d’analyses de biologie médicale les praticiens agréés désignent parmi eux un coordinateur et communiquent son nom à l’agence régionale de l’hospitalisation et à l’Agence de la biomédecine. Le praticien coordinateur est chargé d’organiser la concertation pluridisciplinaire préalable à la mise en œuvre de toute assistance médicale à la procréation.
Personne responsable : la section 7 du code de la santé publique (Art. R. 2142-37 à R. 2142-38) précise les qualifications ainsi que les missions de la personne responsable. Le directeur de l’établissement, de l’organisme ou du laboratoire ou l’administrateur du groupement de coopération sanitaire autorisé adresse au directeur général de l’Agence de la biomédecine ainsi qu’au directeur général de l’Agence régionale de santé une copie de l’acte portant désignation de la personne responsable. Lorsque la personne responsable est remplacée, il communique immédiatement au directeur général de l’agence le nom et la date de prise de fonction de la personne nouvellement désignée.
Correspondant local d’AMP vigilance : la section 8 du code de la santé publique (Art. R2142-47) précise les missions. Les établissements et organismes mentionnés au 2° de l’article R. 2142-42 désignent un correspondant local et son suppléant. Dès leur désignation, l’identité, la qualité, l’expérience et les coordonnées du correspondant local et de son suppléant sont communiquées à l’Agence de la biomédecine par le responsable de la structure dans laquelle le correspondant et son suppléant exercent leurs fonctions.
à remplir et à retourner :
![]() par mail (voie à privilégier) : ampvigilance@biomedecine.fr
par mail (voie à privilégier) : ampvigilance@biomedecine.fr
![]() ou par courrier :
ou par courrier :
Agence de la biomédecine
Pôle sécurité-qualité
1, avenue du stade de France
93212 SAINT-DENIS LA PLAINE CEDEX
Après réception de ces informations, les correspondants locaux d’AMP vigilance sont inscrits dans l’annuaire national des CLA géré par l’Agence de la biomédecine. Leurs identifiants et mots de passe pour accéder à l’outil de télédéclaration « AMP Vigie » leurs sont envoyés par courrier.
En cas de remplacement d’un correspondant local d’AMP vigilance ou bien de modifications de ses coordonnées, une nouvelle fiche d’identification devra être saisie et renvoyée.
La déclaration d’AMPVigilance
Tout incident grave ou effet indésirable doit être déclaré sans délai à l’Agence de la biomédecine via l’outil de télédéclaration « AMP Vigie » en l’absence de référentiels.
En l’absence d’accès à l’application (mot de passe perdu, correspondant local non encore désigné, etc.), une déclaration, dont les données nominatives concernant les patients/couples/donneurs sont rendues anonymes, peut être exceptionnellement envoyée par mail (ampvigilance@biomedecine.fr) ou par courrier (Agence de la biomédecine / Pôle sécurité-qualité, 1 avenue du stade de France 93212 Saint Denis La Plaine Cedex) à l’aide des modèles de fiches ci-dessous :
Pour les professionnels de santé exerçant en ville ou dans un établissement de santé ne disposant pas d’un centre d’AMP, la déclaration peut s’effectuer en passant par le portail des signalements du ministère (https://signalement.social-sante.gouv.fr) ou en se mettant en relation avec le centre d’AMP ayant pris en charge le couple ou le patient afin que ce dernier fasse la déclaration auprès de l’Agence de la biomédecine.
Schéma récapitulatif du circuit de déclaration :
Le rapport annuel du CLA
Le CLA est tenu de rédiger un rapport annuel qu’il devra adresser à l’Agence de la biomédecine avant le 31 mars de l’année n+1. Ce rapport comportera une synthèse de l’ensemble des incidents et effets indésirables déclarés ou surveillés. L’objectif est de présenter les mesures correctives visant à limiter la criticité des événements observés dans son centre que ceux-ci aient fait l’objet de déclaration à l’Agence de la biomédecine ou de mesures de surveillance locale. Afin de faciliter la rédaction du rapport, l’Agence de la biomédecine pré-remplira pour chaque CLA son rapport avec les déclarations qu’il aura effectuées au cours de l’année n.
Le guide pratique d’aide à la mise en place de l’AMP vigilance dans un établissement de santé
Ce document est destiné avant tout aux correspondants locaux d’AMP Vigilance. Il devrait leur permettre de préciser leurs missions, les circuits de signalement et de déclaration des incidents et des effets indésirables ainsi que le rôle des différents acteurs impliqués.
Les rapports annuels d’AMP vigilance
Conformément aux textes réglementaires (décret du 29 novembre 2016), ces rapports annuels sont adressés chaque année par l’ABM au ministre de la santé et à la Commission européenne.
Consulter les anciens rapports annuels d’AMP vigilance (Sélectionner les mots-clés : "AMP + Vigilance + Rapport d’activité")
Les réunions des correspondants locaux d’AMP vigilance
Depuis 2008, une réunion nationale des correspondants locaux d’AMP vigilance se déroule sur une demi-journée et est organisée en marge du congrès de la Fédération Française d’Etude de la Reproduction (FFER) tous les 1 à 2 ans.
Cette réunion a pour objectifs :
![]() d’informer les professionnels et notamment les CLA des actualités du dispositif d’AMP vigilance ;
d’informer les professionnels et notamment les CLA des actualités du dispositif d’AMP vigilance ;
![]() de promouvoir le dispositif en sensibilisant les professionnels à l’intérêt d’intégrer cette vigilance dans leurs pratiques ;
de promouvoir le dispositif en sensibilisant les professionnels à l’intérêt d’intégrer cette vigilance dans leurs pratiques ;
![]() d’organiser un retour et un partage d’expériences entre professionnels.
d’organiser un retour et un partage d’expériences entre professionnels.
Les comptes rendus de ces réunions font l’objet d’une newsletter « AMP Vigilance’infos » qui est ensuite diffusée aux correspondants locaux d’AMP vigilance (cf. Infra).
Lettre d’information « AMP vigilance’ infos »
Sur la base des déclarations réceptionnées dans le dispositif AMP Vigie, l’Agence de la biomédecine publie une lettre d’information « AMP Vigilance’infos ».
Destinée à favoriser le retour d’information vers les professionnels concernés, n’hésitez pas à la relayer et à faire part de vos retours à la direction générale médicale et scientifique - Pôle Sécurité-Qualité : ampvigilance@biomedecine.fr ou Tél : 01 55 93 69 03 ou 64 53
2024
2022
2020
2019
2017
2016
2015
2014
2013
2012
Les mises au point / Recommandations
Ces mises au point / recommandations ont été élaborées suite à la déclaration d’événements indésirables. Elles ont été élaborées selon une méthodologie de consensus d’experts internes et externes et sont susceptibles d’évoluer en fonction de l’état de l’art.
La cartographie des risques en AMP
La gestion d’une salle de cryoconservation
Les syndromes d’hyperstimulation ovarienne sévère (SHOS)
Les déclarations de Syndromes d’Hyperstimulation Ovarienne Sévère (SHOS) représentent environ la moitié des déclarations d’AMP vigilance. De par ses missions dans la promotion de la qualité et de la sécurité des soins, l’Agence de la biomédecine, en 2012, a mis à disposition des professionnels de santé des outils d’évaluation des pratiques professionnelles (EPP) dans la prise en charge des SHOS. L’objectif de ces outils étant de contribuer à la prévention et à une meilleure identification des risques de survenue du syndrome d’HSO.
Le cahier d’observation électronique permettant le recueil des données n’étant plus mis à disposition sur le site eppshos.org, les documents relatifs à cet EPP sont disponibles ci-après.
Erreur d’attribution de gamètes ou d’embryons
RETEX infection post-ponction ovocytaire
Thromboses artérielles et veineuses dans le cadre de l’AMP : recommandations de prévention et de prise en charge – Label HAS
RETEX accident d’anesthésie au cours d’une ponction ovocytaire
Sécurité virale en AMP
Syndrome de Turner
Divers
Textes juridiques
Vos interlocuteurs à L’Agence de la biomédecine
Le Pôle sécurité-qualité, Direction générale médicale et scientifique
![]() Responsable des vigilances : Dr Jacques-Olivier Galdbart
Responsable des vigilances : Dr Jacques-Olivier Galdbart
![]() Référent AMP vigilance : Mme Gaelle Lemardeley
Référent AMP vigilance : Mme Gaelle Lemardeley
Tel : 01 55 93 69 03
Fax : 01 55 93 69 36
Mèl : ampvigilance@biomedecine.fr